Nach der Lektüre und Verarbeitung dieses Artikels sollte die praktizierende medizinische Fachkraft in der Lage sein, die Bedeutung der Begriffe Pharmakokinetik und Pharmakodynamik zu diskutieren; und:
- Vergleichen und kontrastieren Sie Pharmakokinetik und Pharmakodynamik
- Diskutieren Sie die Bedeutung der Kreatinin-Clearance
- Diskutieren Sie die Bedeutung der Halbwertszeit eines Medikaments
- Diskutieren Sie die Bedeutung des P450-Stoffwechselwegs.
Was ist Pharmakologie
Medikamente haben das Ziel, verschiedene Krankheitszustände zu verhindern, zu heilen oder zu kontrollieren. Um dieses Ziel zu erreichen, müssen adäquate Konzentrationen des Medikaments an die Zielgewebe geliefert werden, so dass therapeutische, aber nicht toxische Werte erreicht werden.
Die pharmakologischen und toxikologischen Wirkungen von Medikamenten hängen in erster Linie von ihren Plasmakonzentrationen ab. Folglich muss das medizinische Fachpersonal, das mit Arzneimitteln arbeitet, die Geschwindigkeit des Wirkungseintritts sowie die Intensität und Dauer der Wirkung des Medikaments kennen. Diese wiederum werden durch die folgenden vier grundlegenden Wege der Medikamentenbewegung und -modifikation im Körper gesteuert:
- Absorption
- Verteilung
- Metabolismus
- Ausscheidung, sowie Wirkungseintritt und -dauer
Pharmakokinetik vs. Pharmakodynamik
Die Pharmakokinetik beeinflusst den entschiedenen Verabreichungsweg für ein bestimmtes Medikament, die Menge und Häufigkeit der jeweiligen Dosis sowie die Dosierungsintervalle.
Die Pharmakodynamik hingegen ist die Lehre von der Wirkung eines Medikaments auf einen lebenden Organismus. Dies beinhaltet die pharmakologische Reaktion und ihre Dauer und Größe, die im Verhältnis zur Konzentration des Medikaments an einer aktiven Stelle im Organismus beobachtet wird; d.h. die Untersuchung der Wirkung eines Medikaments und der Wirkmechanismen.
Was Medikamente im Körper bewirken
Die klinische Pharmakokinetik ist die Anwendung pharmakokinetischer und pharmakodynamischer Prinzipien für die sichere und effektive therapeutische Behandlung eines einzelnen Patienten.
Halbwertszeitformel
Der pharmakokinetische Begriff Halbwertszeit (t1/2) bezieht sich auf die Zeit, die vergeht, bis die Hälfte der verabreichten Anfangsdosis eines Medikaments aus dem Körper ausgeschieden ist. Das heißt:
Beispielsweise würde bei einer Medikamentendosis mit einer Halbwertszeit von 12 Stunden nach 24 Stunden noch 25 % des Medikaments im Körper vorhanden sein.
Viele Medikamente werden nach ihrer Halbwertszeit klassifiziert. Zum Beispiel werden die Benzodiazepine wie folgt klassifiziert:
- Ultrakurz wirkend (t1/2 < 6 Stunden): Midazolam, Triazolam
- Kurz wirkend (t1/2 6-12 Stunden): Oxazepam, Temazepam
- Mittelstark wirkend (t1/2 12-24 Stunden): Alprazolam, Bromazepam, Lorazepam
- Lang wirkend (t1/2 > 24 Stunden): Clobazepam, Clonazepam, Diazepam, Flunitrazepam, Nitrazepam
Absorption, Verteilung, Metabolismus und Ausscheidung
Nachfolgend finden Sie eine schematische Darstellung der Absorption, Verteilung, des Metabolismus und der Ausscheidung von Arzneimitteln:
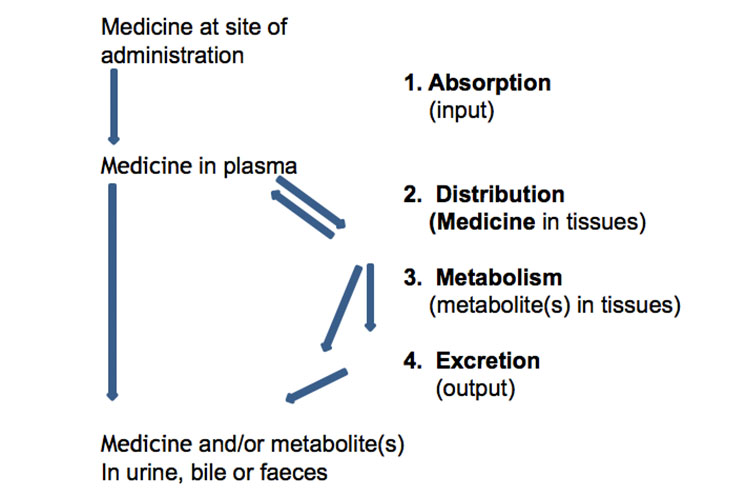
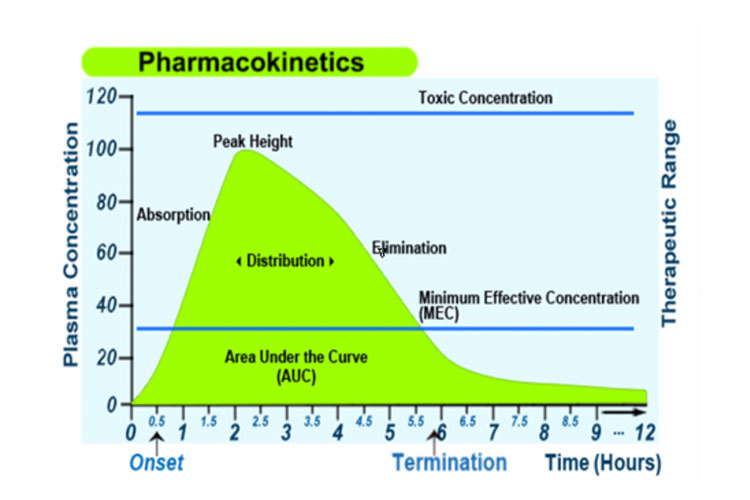
Alternativ stellt die folgende Grafik die Absorption, Verteilung, den Stoffwechsel und die Ausscheidung eines Arzneimittels zusammen mit einigen pharmakokinetischen Begriffen nach einer einzelnen oralen Sofortdosis dar:
Absorption
Die Absorption vom Verabreichungsort ermöglicht den Eintritt des Therapeutikums (entweder direkt oder indirekt) in das Plasma.
Zu den medikamentenbezogenen Faktoren gehören Ionisationszustand, Molekulargewicht, Löslichkeit und Formulierung. Kleine, nicht ionisierte, fettlösliche Medikamente durchdringen die Plasmamembranen am leichtesten.
Verteilung
Nach der Resorption kann das Medikament dann reversibel den Blutkreislauf verlassen und sich in die interstitiellen und intrazellulären Flüssigkeiten verteilen.
Die Permeabilität eines Medikaments wird durch die Blut-Hirn-Schranke, die Blut-Hoden-Schranke und die Blut-Plazenta-Schranke definiert.
Die meisten Medikamente sind bis zu einem gewissen Grad proteingebunden; nur ein ungebundenes Medikament kann seine pharmakologische(n) Wirkung(en) frei entfalten.
Die Depotspeicherung bezieht sich auf lipophile Medikamente, die sich in Fett einlagern, kalziumbindende Medikamente, etc.
Mit dem Altern kommt es zu einer Abnahme der fettfreien Körpermasse und des Gesamtkörperwassergehalts sowie zu einer Zunahme des Gesamtkörperfetts. Dies kann bei einigen Medikamenten zu Veränderungen des Verteilungsvolumens (Vd) führen, was insbesondere bei gebrechlichen älteren Erwachsenen zu unvorhersehbaren Wirkungen führen kann.
Das Verteilungsvolumen ist das Ausmaß, in dem sich ein Medikament aus dem Blutkreislauf heraus und in die Gewebe des Körpers (d. h. den Ort der Arzneimittelrezeptoren) verteilt. Eine Verringerung des Vd führt zu höheren Plasmakonzentrationen für hydrophile Arzneimittel wie Gentamicin, Digoxin und Lithium. Ein höherer Anteil an Körperfett erhöht die Vd für lipophile Arzneimittel wie Diazepam, was zu einer Verlängerung der Plasmahalbwertszeit führt.
Metabolismus
Bevor das Arzneimittel ausgeschieden wird, wird es von der Leber, der Niere oder anderen Stellen metabolisiert. Durch diesen Prozess wird das Medikament polarer (wasserlöslicher), was zu einer Inaktivierung des Medikaments und seiner Ausscheidung führen kann.
Metaboliten können mehr oder weniger (prodrug) aktiv sein als das Muttermedikament. Die Leber ist die Hauptquelle dieser Enzyme (P450-Enzyme), obwohl sie auch im Magen-Darm-Trakt, im Herz, in der Lunge, im Gehirn und in den Nieren vorkommen können.
1. Phase-I-Reaktionen (nicht-synthetisch)
Bei den Phase-I-Reaktionen (nicht-synthetisch) handelt es sich um geringfügige strukturelle Veränderungen der Ausgangsstruktur durch Oxidation, Reduktion oder Hydrolyse, um kleinere, wasserlöslichere Metaboliten zu erzeugen. Diese werden überwiegend von den Cytochrom-P450-Enzymen verarbeitet.
Phase-I-Reaktionen bieten häufig einen „Griff“ für weitere Modifikationen durch nachfolgende Phase-II-Reaktionen.
2. Phase-II-Reaktionen (synthetisch)
Bei Phase-II-Reaktionen (synthetisch) wird ein wasserlösliches körpereigenes Molekül wie Glucuronsäure, Sulfat oder Glutathion an eine Chemikalie (Ausgangssubstanz und/oder Phase-I-Metabolit) gekoppelt, um die Ausscheidung zu erleichtern.
Die häufigsten Ursachen für Arzneimittel-zu-Arzneimittel-Wechselwirkungen liegen in der Pharmakokinetik, insbesondere im Stoffwechsel. Diese werden als Cytochrom-P450-Interaktionen bezeichnet.
Eine große Anzahl klinisch wichtiger Interaktionen entsteht durch die Hemmung oder Induktion von Substraten (Arzneimittel, die maßgeblich von den gegebenen Enzymen verstoffwechselt werden).
- Inhibitoren sind Verbindungen, die generell in der Lage sind, den Stoffwechsel der verschiedenen Substrate zu hemmen. Infolgedessen kann die Verabreichung des Inhibitors zu einer erhöhten Plasmakonzentration des Substrats führen. Zum Beispiel hemmt Ciprofloxacin das CYP3A4-Enzym, das Clozapin metabolisiert, was zu einer Toxizität von Clozapin führen kann.
- Induktoren des angegebenen P450 haben die Fähigkeit, die Aktivität des bezeichneten Enzyms zu erhöhen und somit die Plasmakonzentrationen der aufgeführten Substrate zu verringern. Zum Beispiel induziert Carbamazepin den Metabolismus von Cyclosporin über das CYP3A4-Enzym, was zu einer Verringerung der Plasmaspiegel von Cyclosporin und damit zu einem Verlust der Wirksamkeit führt.

Ausscheidung
Medikamente und ihre Metaboliten werden mit dem Urin aus dem Körper entfernt, Galle und/oder Fäkalien ausgeschieden. In manchen Fällen müssen sie erst zu besser wasserlöslichen Bestandteilen verstoffwechselt werden (Beispiele sind Amiodaron, Amitriptylin, Amlodipin, Amphotericin B, Aripiprazol, Aspirin, Atomoxetin, Atorvastatin, Azithromycin, Felodipin usw.)
Andere werden unverändert oder relativ unverändert ausgeschieden (Penicilline, Cephalosporine, Aminoglykoside, Ganciclovir, Milrinon, Oseltamivir, Risedronat, Vareniclin usw.).
Die wichtigsten an der Ausscheidung beteiligten Prozesse sind die glomeruläre Filtration, die tubuläre Sekretion und die tubuläre Reabsorption.
Die tubuläre Ausscheidung und Rückresorption kann auch durch Medikamente beeinflusst werden, die den Urin entweder saurer (Ammoniumchlorid, hohe Dosen von Vitamin C) oder alkalischer machen (Harnalkalisierer wie Citralite®, Citravescent®, Uracol®, Ural® , die alle den Urin-pH auf etwa pH 9 anheben).
Kreatinin und Kreatinin-Clearance
Kreatinin (Normalbereich: Frauen 50-110 Mikromol /L; Männer 60-120 Mikromol /L) ist das Stoffwechselprodukt des Muskelstoffwechsels. Sein Spiegel spiegelt sowohl die Muskelmasse als auch die Nierenfunktion wider, da Kreatinin überwiegend durch glomeruläre Filtration über die Niere ausgeschieden wird.
Niedrige Werte können auf Proteinmangel, Lebererkrankungen oder Schwangerschaft hinweisen, während hohe Werte bei Nierenversagen, Muskeldegeneration und den Auswirkungen einiger Medikamente, die die Nierensekretion blockieren (z. B. Cimetidin und Triglycerin), zu beobachten sind.
Kreatinin-Clearance (CrCl)
Die Kreatinin-Clearance (CrCl) ist eine Schätzung der glomerulären Filtrationsrate (GFR), die ein direktes Maß für die Nierenfunktion ist. Die CrCl kann mit Hilfe der Cockroft-Gault-Gleichung berechnet werden:
(Bei Frauen mit 0.85, um das geringere Verhältnis von Muskeln zu idealem Körpergewicht im Vergleich zu Männern zu berücksichtigen.)
- Ideales Körpergewicht für Männer = 50 kg + 0,9 kg/jeder cm über 152 cm.
- Ideales Körpergewicht für Frauen = 45,5 kg + 0,9 kg/jeder cm über 152 cm.
(In vielen Medikamentenmonographien werden Dosis und Dosierungshäufigkeit eines Medikaments entsprechend der Kreatinin-Clearance angegeben.)
eGFR
eGFR wird von Pathologielaboren als geschätzter Indikator der Nierenfunktion verwendet. (eGFR hat Einschränkungen, und für die Medikamentendosierung bei einer Nierenfunktion von <60mL/min ist die Cockcroft-Gault-Gleichung vorzuziehen.)
Bioverfügbarkeit
Die Bioverfügbarkeit ist eine Unterkategorie der Absorption und ist der Anteil einer verabreichten Dosis eines unveränderten Medikaments, der den systemischen Kreislauf erreicht. Wenn ein Medikament intravenös verabreicht wird, beträgt seine (absolute) Bioverfügbarkeit definitionsgemäß 100 %.
Die Wirksamkeit eines Medikaments ist eine Funktion der Rate und des Ausmaßes der Absorption. Die relative Bioverfügbarkeit wird als Verhältnis (%) zwischen der Bioverfügbarkeit aus einer oralen Darreichungsform (oder einer anderen nicht-IV-Dosis) relativ zu einer IV-Dosis abgeleitet.
- Bryant, B & Knights, K 2015, Pharmacology for Health Professionals, 4th edn, Mosby Elsevier, Sydney, Australia
- eMIMs 2016, CMPMedica, https://www.mimsonline.com.au/Login/Login.aspx?ReturnUrl=%2fdefault.aspx
- Harris, P, Nagy, S & Vardaxis, N 2014, Mosby’s Dictionary of Medicine, Nursing and Health Professions, 3. edn, Elsevier, Australien.
- Neal, M J 2015, Medical Pharmacology at a Glance, 8th edn, Blackwell Science, Oxford.
- Rossi, S 2016, Australian Medicines Handbook (AMH), https://amhonline.amh.net.au/auth