Po przeczytaniu i zapoznaniu się z tym artykułem, praktykujący pracownik służby zdrowia powinien być w stanie omówić znaczenie terminów farmakokinetyka i farmakodynamika; oraz:
- Porównać i skontrastować farmakokinetykę i farmakodynamikę
- Przedstawić znaczenie klirensu kreatyniny
- Przedstawić znaczenie okresu półtrwania leku
- Przedstawić znaczenie szlaku metabolicznego P450.
Co to jest farmakologia
Leki mają za zadanie zapobiegać, leczyć lub kontrolować różne stany chorobowe. Aby osiągnąć ten cel, odpowiednie stężenie leku musi być dostarczone do tkanek docelowych, tak aby uzyskać poziom terapeutyczny, ale nietoksyczny.
Farmakologiczne i toksykologiczne działania leków są przede wszystkim związane z ich stężeniem w osoczu. W związku z tym pracownicy służby zdrowia, którzy pracują z lekami, muszą rozpoznać szybkość początku działania leku, jak również intensywność i czas trwania jego efektu. Te z kolei są kontrolowane przez następujące cztery podstawowe szlaki przemieszczania się i modyfikacji leków w organizmie:
- Wchłanianie
- Dystrybucja
- Metabolizm
- Wydalanie, jak również początek i czas trwania działania
Farmakokinetyka a farmakodynamika
Farmakokinetyka wpływa na wybraną drogę podania konkretnego leku, ilość i częstotliwość każdej dawki oraz odstępy między dawkami.
Farmakodynamika, z drugiej strony, jest nauką o tym, jak lek działa na żywy organizm. Obejmuje ona odpowiedź farmakologiczną oraz czas jej trwania i wielkość obserwowaną w stosunku do stężenia leku w miejscu aktywnym w organizmie; tj. badanie efektu działania leku i mechanizmów działania.
Co leki robią z ciałem
Farmakokinetyka kliniczna to zastosowanie zasad farmakokinetycznych i farmakodynamicznych do bezpiecznego i skutecznego postępowania terapeutycznego z indywidualnym pacjentem.
Wzór na okres półtrwania
Termin farmakokinetyczny okres półtrwania (t1/2) odnosi się do czasu potrzebnego na wyeliminowanie z organizmu połowy początkowej dawki podanego leku. Czyli:
Na przykład, dawka leku o okresie półtrwania 12 godzin spowoduje, że 25% leku pozostanie w organizmie po 24 godzinach.
Wiele leków jest klasyfikowanych pod względem ich okresów półtrwania. Na przykład, benzodiazepiny są klasyfikowane pod względem:
- Niezwykle krótko działające (t1/2 < 6 godzin): midazolam, triazolam
- Krótko działające (t1/2 6-12 godzin): oksazepam, temazepam
- Średnio działające (t1/2 12-24 godz.): alprazolam, bromazepam, lorazepam
- Długo działające (t1/2 > 24 godz.): klobazepam, klonazepam, diazepam, flunitrazepam, nitrazepam
Wchłanianie, dystrybucja, metabolizm i wydalanie
Poniżej przedstawiono schemat wchłaniania, dystrybucji, metabolizmu i wydalania leków:
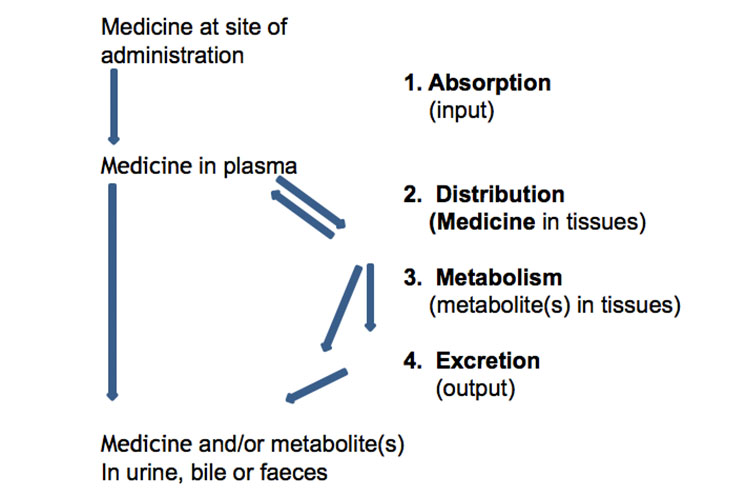
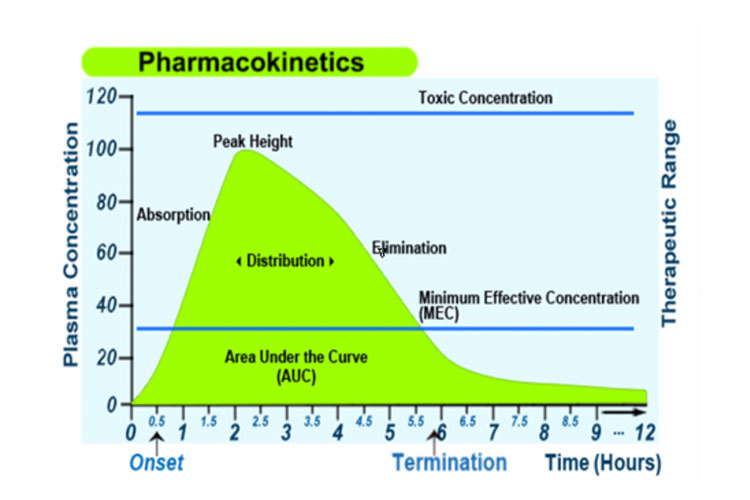
Alternatywnie, następujący wykres przedstawia wchłanianie, dystrybucję, metabolizm i wydalanie leku, wraz z niektórymi terminami farmakokinetycznymi, po podaniu pojedynczej doustnej dawki o natychmiastowym uwalnianiu:
Absorpcja
Absorpcja z miejsca podania umożliwia wprowadzenie środka leczniczego (bezpośrednio lub pośrednio) do osocza.
Czynniki związane z medycyną obejmują stan jonizacji, masę cząsteczkową, rozpuszczalność i postać. Małe, niezjonizowane, rozpuszczalne w lipidach leki najłatwiej przenikają przez błony osocza.
Dystrybucja
Po wchłonięciu lek może w sposób odwracalny opuścić krwiobieg i rozprowadzić się do płynów śródmiąższowych i wewnątrzkomórkowych.
Przepuszczalność leku jest określona przez barierę krew-mózg, barierę krew-test i barierę krew-placenta.
Większość leków jest w pewnym stopniu związana z białkami; tylko lek niezwiązany może swobodnie wykonywać swoje działanie(a) farmakologiczne.
Przechowywanie w depozycie odnosi się do leków lipofilnych, które przechowują się w tłuszczu, leków wiążących wapń itp.
Wraz ze starzeniem się dochodzi do zmniejszenia beztłuszczowej masy ciała i całkowitej zawartości wody w organizmie oraz zwiększenia całkowitej zawartości tkanki tłuszczowej. Może to prowadzić do zmian w objętości dystrybucji (Vd) niektórych leków, powodując nieprzewidywalne skutki, szczególnie u słabych osób w podeszłym wieku.
Objętość dystrybucji to stopień, w jakim lek rozprowadza się poza krwiobiegiem i do tkanek organizmu (tj. do miejsca receptorów leku). Zmniejszenie Vd będzie skutkowało wyższym stężeniem w osoczu leków hydrofilnych, takich jak gentamycyna, digoksyna i lit. Większy odsetek tkanki tłuszczowej w organizmie zwiększy Vd dla leków lipofilnych, takich jak diazepam, powodując wydłużenie okresu półtrwania w osoczu.
Metabolizm
Przed wydaleniem lek jest metabolizowany przez wątrobę, nerki lub inne miejsca. Jest to proces powodujący, że lek staje się bardziej polarny (bardziej rozpuszczalny w wodzie), co może prowadzić do inaktywacji leku i jego wydalania.
Metabolity mogą być bardziej lub mniej (prolek) aktywne niż lek macierzysty. Wątroba jest głównym źródłem tych enzymów (enzymy P450), chociaż mogą one być obecne w przewodzie pokarmowym, sercu, płucach, mózgu i nerkach.
1. Reakcje I fazy (niesyntetyczne)
Reakcje I fazy (niesyntetyczne) obejmują niewielkie modyfikacje strukturalne struktury macierzystej poprzez utlenianie, redukcję lub hydrolizę w celu wytworzenia mniejszych, bardziej rozpuszczalnych w wodzie metabolitów. Są one głównie obsługiwane przez enzymy cytochromu P450.
Reakcje fazy I często stanowią „uchwyt” dla dalszych modyfikacji w kolejnych reakcjach fazy II.
2. Reakcje fazy II (syntetyczne)
Reakcje fazy II (syntetyczne) polegają na sprzężeniu rozpuszczalnej w wodzie cząsteczki endogennej, takiej jak kwas glukuronowy, siarczan lub glutation, z substancją chemiczną (związkiem macierzystym i/lub metabolitem fazy I) w celu ułatwienia wydalania.
Najczęstszą przyczyną interakcji między lekami jest farmakokinetyka, zwłaszcza metaboliczna. Są one znane jako interakcje cytochromu P450.
Duża liczba klinicznie ważnych interakcji wynika z hamowania lub indukcji substratów (leków, które są w znacznym stopniu metabolizowane przez dane enzymy).
- Inhibitory to związki, które są ogólnie zdolne do hamowania metabolizmu różnych substratów. W związku z tym podanie inhibitora może prowadzić do zwiększenia stężenia substratu w osoczu. Na przykład, cyprofloksacyna hamuje enzym CYP3A4, który metabolizuje klozapinę, co może prowadzić do toksyczności klozapiny.
- Induktory określonych P450 mają zdolność do zwiększania aktywności wskazanego enzymu, a tym samym zmniejszania stężenia wymienionych substratów w osoczu. Na przykład karbamazepina indukuje metabolizm cyklosporyny za pośrednictwem enzymu CYP3A4, co prowadzi do zmniejszenia stężenia cyklosporyny w osoczu, a tym samym utraty skuteczności.

Wydalanie
Medycyny i ich metabolity są usuwane z organizmu z moczem, żółci i/lub kale. W niektórych przypadkach najpierw muszą być metabolizowane do bardziej rozpuszczalnych w wodzie cząsteczek (przykłady obejmują amiodaron, amitryptylinę, amlodypinę, amfoterycynę B, aripiprazol, aspirynę, atomoksetynę, atorwastatynę, azytromycynę, felodypinę itp.)
Inne są wydalane w postaci niezmienionej lub względnie niezmienionej (penicyliny, cefalosporyny, aminoglikozydy, gancyklowir, milrinon, oseltamiwir, risedronian, wareniklina itp.).
Główne procesy biorące udział w wydalaniu to filtracja kłębuszkowa, wydzielanie kanalikowe i reabsorpcja kanalikowa.
Na wydalanie kanalikowe i reabsorpcję mogą również wpływać leki, które albo czynią mocz bardziej kwaśnym (chlorek amonu, duże dawki witaminy C), albo bardziej zasadowym (środki alkalizujące mocz, takie jak Citralite®, Citravescent®, Uracol®, Ural® , które podnoszą pH moczu do około pH 9).
Kreatynina i klirens kreatyniny
Kreatynina (zakres normy: kobiety 50-110 mikromoli /L; mężczyźni 60-120 mikromoli /L) jest produktem przemiany materii w mięśniach. Jej poziom jest odzwierciedleniem zarówno masy mięśniowej, jak i funkcji nerek, ponieważ kreatynina jest eliminowana głównie poprzez filtrację kłębuszkową przez nerki.
Niski poziom może wskazywać na głodzenie się białkiem, choroby wątroby lub ciążę, natomiast wysoki poziom jest obserwowany w niewydolności nerek, zwyrodnieniu mięśni i działaniu niektórych leków, które blokują wydzielanie nerkowe (np.np. cymetydyna i trimetoprim).
Klirens kreatyniny (CrCl)
Klirens kreatyniny (CrCl) jest szacunkowym wskaźnikiem filtracji kłębuszkowej (GFR), który jest bezpośrednią miarą funkcji nerek. CrCl można obliczyć za pomocą równania Cockrofta-Gaulta:
(W przypadku kobiet, pomnożyć przez 0.85, aby uwzględnić zmniejszony stosunek masy mięśniowej do idealnej masy ciała w porównaniu z mężczyznami.)
- Idealna Waga Ciała dla Mężczyzn = 50 kg + 0.9 kg/każdy cm powyżej 152 cm.
- Idealna Waga Ciała dla Kobiet = 45.5 kg + 0.9 kg/każdy cm powyżej 152 cm.
(Wiele monografii leków wskazuje dawkę i częstotliwość dawkowania leku w zależności od klirensu kreatyniny.)
eGFR
eGFR jest używany przez laboratoria patologiczne jako szacunkowy wskaźnik funkcji nerek. (eGFR ma ograniczenia, a do dawkowania leków, gdy czynność nerek wynosi <60mL/min, preferowane jest równanie Cockcrofta-Gaulta.)
Dostępność biologiczna
Dostępność biologiczna jest podkategorią wchłaniania i jest frakcją podanej dawki niezmienionego leku, która dociera do krążenia ogólnoustrojowego. Z definicji, kiedy lek jest podawany dożylnie, jego (bezwzględna) biodostępność wynosi 100%.
Skuteczność leku jest funkcją szybkości i zakresu wchłaniania. Biodostępność względna jest uzyskiwana jako stosunek (%) pomiędzy biodostępnością z formy doustnej (lub innej dawki nie dożylnej) w stosunku do dawki dożylnej.
- Bryant, B & Knights, K 2015, Pharmacology for Health Professionals, 4th edn, Mosby Elsevier, Sydney, Australia
- eMIMs 2016, CMPMedica, https://www.mimsonline.com.au/Login/Login.aspx?ReturnUrl=%2fdefault.aspx
- Harris, P, Nagy, S & Vardaxis, N 2014, Mosby’s Dictionary of Medicine, Nursing and Health Professions, 3rd edn, Elsevier, Australia.
- Neal, M J 2015, Medical Pharmacology at a Glance, 8th edn, Blackwell Science, Oxford.
- Rossi, S 2016, Australian Medicines Handbook (AMH), https://amhonline.amh.net.au/auth
.