To są notatki z wykładu 11 kursu Biologii Komórki Harvard Extension.
W tym wykładzie zostaną omówione dwa różne sposoby, w jakie komórki mogą umierać: apoptoza (zaprogramowana śmierć komórki) i nekroza (nieplanowana śmierć komórki). Łatwo jest rozróżnić te dwa sposoby morfologicznie pod mikroskopem, jak pokazano na tym obrazku z Wikimedia Commons:
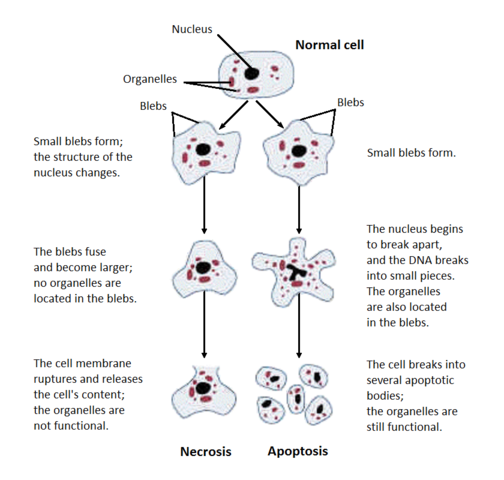
Nekroza
Nekroza ma miejsce wtedy, gdy komórki umierają przypadkowo z powodu, powiedzmy, urazu (np. ukąszenie jadowitego pająka), lub braku składników odżywczych (np. brak dopływu krwi). Nekroza zaczyna się od obrzęku komórki, chromatyna zostaje strawiona, błony plazmy i organelli zostają przerwane, ER wakuolizuje, organelle rozpadają się całkowicie i w końcu komórka ulega rozpadowi, wypluwając swoją zawartość wewnątrzkomórkową i wywołując odpowiedź immunologiczną (zapalenie).
Apoptoza
Apoptoza może stanowić samobójstwo lub zabójstwo komórki. Komórki popełniają samobójstwo, gdy brakuje im jakiegokolwiek sygnału przeżycia w postaci czynników troficznych lub gdy wykryją rozległe uszkodzenia DNA we własnym jądrze. Komórki będą mordować inne komórki, aby usunąć niepotrzebne komórki lub wyeliminować potencjalnie atakujące komórki odpornościowe.
Każdy z tych procesów stanowi zaprogramowaną śmierć komórki. Podczas rozwoju embrionalnego ludzie mają pajęcze ręce, stopy i ogony; komórki, które tworzą te części później apoptują. Apoptoza przebiega również nieustannie w wielu tkankach, w tym w jelitach.
Oto wspaniały obraz apoptozy z Wikimedia Commons (czytaj od lewej do prawej, od góry do dołu) dzięki Egelbergowi:
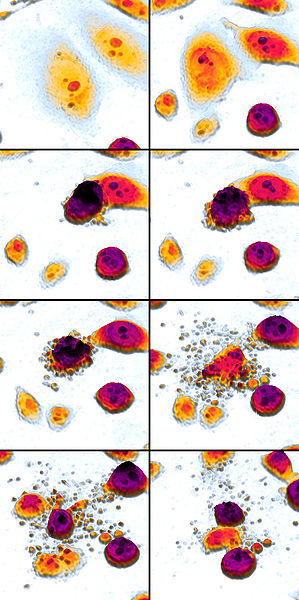
Główne etapy apoptozy:
- Komórka się kurczy
- Komórka się fragmentuje
- Cytoszkielet się zapada
- Opuszka jądrowa się rozpada
- Komórki uwalniają ciała apoptotyczne
Niezwykle nieobecne na tej liście jest 'wysyłanie sygnału'.' Komórki apoptotyczne nie wysyłają żadnych sygnałów, z jednym wyjątkiem: uwalniają ciała apoptotyczne i „białka pochłaniające”, aby nakłonić inne komórki (komórki fagocytujące) do pochłonięcia ciał apoptotycznych i rozbicia ich w lizosomach, ale nie jest to zbyt silna odpowiedź immunologiczna.
Białka ważne w apoptozie:
- „białka zabójcze”: kaspazy (omówione szczegółowo poniżej).
- „białka niszczące”, które trawią DNA, fragmentują komórkę i rozbijają cytoszkielet
- „białka pochłaniające”, które wywołują i promują fagocytozę przez inne komórki
C. elegans jest głównym organizmem modelowym dla zrozumienia apoptozy, zarówno przez genetykę w przód, jak i w tył. Genetyka postępu polega na obserwacji fenotypu, a następnie określeniu, który gen go wywołuje; genetyka odwrotna polega na wprowadzeniu mutacji do znanego genu w celu sprawdzenia, jaki fenotyp powstaje.
Kluczowy szlak apoptozy u C. elegans jest pokazany na tym rysunku Google, który stworzyłem:
Oto wyjaśnienie, jak każde z tych białek wykonuje swoją pracę, od dołu do góry:
Są:
- CED-3 pociąga za spust, aktywując białka apoptotyczne, które niszczą komórkę. (U ssaków odpowiednikiem CED-3 jest kaspaza 9, która rozszczepia się, a tym samym aktywuje kaspazę 3, która z kolei niszczy komórkę.)
- CED-4 aktywuje CED-3.
- CED-9 wiąże się z CED-4, zapobiegając jego aktywacji
- EGL-1 jest aktywowany transkrypcyjnie w odpowiedzi na sygnały śmierci i katalizuje uwolnienie CED-4 z CED-9.
Zauważ, że w tym systemie nie ma żadnej odporności – to pojedyncze punkty awarii na całej drodze. Jeśli CED-3 jest znokautowany, nie może wystąpić apoptoza. Jeśli CED-4 jest znokautowany, nie może wystąpić apoptoza. Jeśli CED-9 zostanie znokautowany, każda komórka w robaku ulegnie apoptozie. Jeśli EGL-1 zostanie znokautowany, apoptoza nie może wystąpić. Zauważ, że kolejność strzałek na powyższym diagramie odzwierciedla przepływ informacji w systemie. Na przykład, jeśli znokautowane zostaną zarówno EGL-1, jak i CED-9, będzie tak samo, jak w przypadku znokautowania samego CED-9: każda komórka ulegnie apoptozie.
W ssakach apoptoza jest regulowana głównie przez kaspazy (proteazy cysteinowo-asparaginowe). Cały szlak kaspaz jest regulowany potranslacyjnie: kaspazy są zawsze obecne w formie nieaktywnej (zwanej procaspazą, zawierającą prodomenę, która zawiera domenę rekrutacyjną kaspaz (CARD)) i mogą być aktywowane przez rozszczepienie. Pozwala to na bardzo szybką reakcję w przypadku konieczności samobójstwa komórki. Aby doszło do apoptozy, kaspazy inicjujące muszą ulec rozszczepieniu i dimeryzacji. W ten sposób aktywowane, muszą rozszczepić kaspazy efektorowe (zwane pro-kaspazami), uruchamiając „kaskadę kaspaz”. Powoduje to zwiększenie liczby aktywowanych kaspaz w komórce. Kaspazy efektorowe mają wiele celów, w tym blaszkę jądrową i cytoszkielet.
Istnieją zarówno kaspazy pro-odżywcze, jak i pro-apoptotyczne, i mają one wiele wspólnych domen. Kaspazy pro-odżywcze mają BH1, 2, 3 i 4; kaspazy pro-apoptotyczne mają albo BH1, 2 i 3, albo tylko BH3.
Białka inhibitora apoptozy (IAP) powstrzymują zarówno kaspazy inicjujące, jak i efektorowe. Każde z nich posiada domenę wiążącą cynk, która łączy się bezpośrednio z kaspazami, hamując ich aktywność.
Jest jednak również białko mitochondrialne SMAC i DIABLO, które hamują inhibitory. Po uszkodzeniu mitochondrium są one uwalniane i wiążą IAPs, uwalniając kaspazy, aby mogły wywołać apoptozę. Inny zbiór białek mitochondrialnych zwany Htra2/Omi, czynnik indukujący apoptozę (AIF) i endonukleaza G mogą również zostać uwolnione i rozszczepić IAPs. AIF powoduje również kondensację chromosomów i fragmentację DNA niezależnie od kaspaz.
Indywidualnie, mitochondria są centralnymi regulatorami apoptozy. W proces ten zaangażowane są białka zewnętrznej błony mitochondrialnej Bcl-2, białka BH3-only oraz Bax: Bax może utworzyć por w błonie, aby umożliwić cytochromowi c, normalnie znajdującemu się w przestrzeni międzybłonowej, wydostanie się do cytozolu. Monomery Bax przemieszczają się z cytoplazmy do zewnętrznej błony mitochondrialnej, gdzie oligomeryzują i umożliwiają napływ jonów przez błonę. Zostało to również wykazane w eksperymentach in vitro, w których można wykazać, że pęcherzyki zbudowane z zewnętrznych memrbanów mitochondrialnych ulegają permeabilizacji w obecności Bax. Obecnie nie wiadomo, dlaczego ten napływ jonów prowadzi do uwolnienia cytochromu c.
Bcl-2 zapobiega uwolnieniu cytochromu c, blokując w ten sposób apoptozę. Bcl-2 był pierwszym genem apoptozy u ssaków, który został sklonowany. W niektórych chłoniakach ulega on translokacji do pozycji pod silniejszym promotorem, powodując nadekspresję, która uniemożliwia komórce nowotworowej apoptozę. Zobacz też zły & bid.
Po uwolnieniu cytochromu c wiąże się on z Apaf-1 (czynnik aktywujący proteazy apoptotyczne), powodując hydrolizę ATP, z którym jest zwykle związany, co powoduje zmianę konformacyjną, która aktywuje Apaf-1 i uruchamia kaskadę kaspaz. Apaf-1 tworzy heptamer w kształcie dysku zwany „kołem śmierci” lub apoptosomem, który aktywuje kaspazy (Wikimedia Commons image by Org1012):
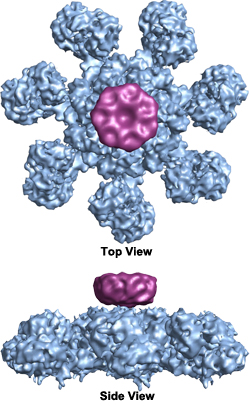
Gdy obecny jest czynnik troficzny, receptor aktywuje PI3K, który aktywuje PKB/Akt, który fosforyluje Bad. p-Bad jest następnie zatrzymywany w cytozolu przez 14-3-3, co zapobiega hamowaniu Bcl-2 przez p-Bad. W ten sposób apoptoza jest uniemożliwiona.
Faktory troficzne są przykładem sygnału zewnątrzkomórkowego, który promuje przeżycie. Istnieją również sygnały zewnątrzpochodne, które promują śmierć (jest to zabójstwo komórki). Czynnik martwicy nowotworów (TNF-alfa) jest uwalniany przez makrofagi w celu wywołania śmierci komórki poprzez wiązanie się z „receptorami śmierci”. Receptory śmierci mają pojedynczą domenę transmembranową. Muszą się one trimeryzować, aby aktywować FADD (Fas-associated death domain). Służą one jako adaptery dla kaspaz-8 i -10 i tworzą kompleks sygnałowy indukujący śmierć (DISC), który może zainicjować kaskadę kaspaz. Chociaż cały ten proces zachodzi niezależnie od mitochondriów, może on również aktywować (?) t-Bid, prowadząc do mitochondrialnego sygnału apoptozy.
Komórki mogą stać się odporne na morderstwo poprzez ekspresję receptorów typu decoy, które mają tylko domenę wiążącą „ligand śmierci” i nie mają aktywnej domeny cytozolowej. Zdarza się to czasami normalnie w komórkach zwierzęcych, ale jest to również sztuczka, którą stosują niektóre wirusy – kodują one białka receptorów typu decoy, aby uchronić komórki gospodarza przed atakiem immunologicznym.
TNF-alfa zwykle promuje śmierć, ale może również promować przeżycie w niektórych typach komórek poprzez aktywację NF-κB. Czasami komórki używają receptorów wabików, aby promować odpowiedź zapalną zamiast śmierci.
P53 jest kluczowym regulatorem odpowiedzi na uszkodzenia DNA i może promować naprawę DNA, apoptozę lub zatrzymanie cyklu komórkowego. Robi to poprzez wiązanie się z promotorami genów docelowych. Nadal nie jest jasne, co decyduje o tym, kiedy p53 będzie indukować zatrzymanie cyklu komórkowego, a kiedy apoptozę.
Metody eksperymentalne
Komórki apoptotyczne wykazują szczególną sygnaturę chemiczną. Jedna z nich polega na tym, że endonukleaza rozszczepia DNA na fragmenty w regionach łącznikowych między nukleosomami, a powstałe fragmenty tworzą drabinkę, gdy są prowadzone na żelu. Inną metodą jest barwienie TUNEL (Terminal deoxynucleotide transferase dUTP Nick End Labeling). Polega to na dodaniu enzymu Tdt i BrdU, który Tdt doda do końców rozciętego DNA. Po daniu mu szansy na wykonanie tej czynności zmywa się nadmiar BrdU, a następnie stosuje przeciwciało przeciwko BrdU. Jeszcze inna metoda polega na tym, że fosfatydyloseryna (PS) normalnie znajduje się w cytozolowym listku błony plazmatycznej; podczas apoptozy przesuwa się do listka egzoplazmatycznego, gdzie służy jako sygnał do żądania od innych komórek, aby fagocytowały umierającą komórkę. Fluorescencyjnie znakowane białko aneksyny V może znakować PS na zewnątrz komórek apoptotycznych.
Dwuniciowe DNA nie może przedostać się przez błonę plazmatyczną nienaruszonych komórek – a to oznacza zdrowe komórki i komórki apoptotyczne. Jeśli jednak się wydostanie, jest to oznaką martwicy. Tak więc można barwić aneksyną V egzoplazmatyczne PS i 7-AAD dsDNA; komórki apoptotyczne to te, które są pozytywne dla aneksyny V, ale negatywne dla 7-AAD.
Podsumowując, oto niepokojący film o apoptozie:
Związek z PrP
Ogólnie, w naturze, komórki albo umierają przez apoptozę, nekrozę, albo przez autofagię (co w tym przypadku oznacza, że zostają pochłonięte w całości przez inne komórki). Nie ma naprawdę żadnych innych sposobów, aby przejść. Jedną z wielu tajemniczych rzeczy dotyczących chorób prionowych jest sposób, w jaki neurony umierają w zakażonym prionami mózgu – nie wydają się one oczywiście podążać żadną z tych ścieżek. Oto cytat z doskonałej pracy na temat toksycznych mechanizmów choroby prionowej :
Rozszczepienie kaspazy 12 nastąpiło w 10wpi, po wzroście ekspresji CHOP… zbiegło się z początkiem utraty neuronów… jednak dokładny mechanizm efektorowy śmierci neuronów jest niejasny: nie znaleźliśmy ani apoptozy, ani autofagii, ani martwicy w badaniu plastrów hipokampa… i ani delecja Bax, ani nadekspresja Bcl-2, ani niedobór kaspazy 12 nie są neuroprotekcyjne w chorobie prionowej.
Oprócz jej własnych dowodów, Moreno przytacza badania infekcji prionowej w modelach mysich z usuniętym Bax (białko pro-apoptotyczne) lub nadekspresją Bcl-2 (białko anty-apoptotyczne) – dwa różne sposoby blokowania apoptozy. Żaden z tych modeli myszy nie miał żadnego opóźnienia lub poprawy choroby prionowej . Inne białko apoptotyczne, kaspaza-12, ulega proteolitycznemu przetwarzaniu podczas infekcji prionowej, ale usunięcie kaspazy-12 również nie zmieniło przebiegu choroby prionowej .