Na het lezen en verwerken van dit artikel zou de beroepsbeoefenaar in de gezondheidszorg in staat moeten zijn om de betekenis van de termen farmacokinetica en farmacodynamica te bespreken; en:
- Farmacokinetiek en farmacodynamiek kunnen vergelijken en contrasteren
- Het belang van creatinineklaring kunnen bespreken
- Het belang van de halfwaardetijd van een geneesmiddel kunnen bespreken
- Het belang van de P450-metabole route kunnen bespreken.
Wat is farmacologie
Medicijnen hebben tot doel verschillende ziektetoestanden te voorkomen, te genezen of te beheersen. Om dit doel te bereiken moeten adequate concentraties van het geneesmiddel aan de doelweefsels worden afgegeven, zodat therapeutische, maar niet-toxische niveaus worden verkregen.
De farmacologische en toxicologische werking van geneesmiddelen houdt voornamelijk verband met hun plasmaconcentraties. Daarom moeten beroepsbeoefenaren in de gezondheidszorg die met geneesmiddelen werken, de snelheid waarmee het geneesmiddel begint te werken en de intensiteit en duur van het effect ervan onderkennen. Deze worden op hun beurt gecontroleerd door de volgende vier fundamentele paden voor de verplaatsing en wijziging van geneesmiddelen in het lichaam:
- Absorptie
- Distributie
- Metabolisme
- Excretie, evenals het begin en de duur van de werking
Farmacokinetiek versus farmacodynamiek
Farmacokinetiek beïnvloedt de gekozen toedieningsweg voor een specifiek geneesmiddel, de hoeveelheid en frequentie van elke dosis en de doseringsintervallen.
Pharmacodynamiek daarentegen is de studie van de werking van een geneesmiddel op een levend organisme. Dit omvat de farmacologische respons en de waargenomen duur en omvang ervan, in verhouding tot de concentratie van het geneesmiddel op een actieve plaats in het organisme; d.w.z. de studie van de werking van een geneesmiddel en de werkingsmechanismen.
Wat geneesmiddelen met het lichaam doen
Klinische farmacokinetiek is de toepassing van farmacokinetische en farmacodynamische principes op de veilige en effectieve therapeutische behandeling van een individuele patiënt.
Half-life Formula
De farmacokinetische term halfwaardetijd (t1/2) verwijst naar de tijd die nodig is om de helft van de oorspronkelijk toegediende dosis geneesmiddel uit het lichaam te elimineren. Dat is:
Bij een dosis geneesmiddel met een halfwaardetijd van 12 uur zou bijvoorbeeld na 24 uur nog 25% van het geneesmiddel in het lichaam aanwezig zijn.
Veel geneesmiddelen zijn geclassificeerd in termen van hun halfwaardetijd. Zo worden de benzodiazepinen ingedeeld in:
- Ultra-kortwerkend (t1/2 < 6 uur): midazolam, triazolam
- Kortwerkend (t1/2 6-12 uur): oxazepam, temazepam
- Middel-werkend (t1/2 12-24 uur): alprazolam, bromazepam, lorazepam
- Lang-werkend (t1/2 > 24 uur): clobazepam, clonazepam, diazepam, flunitrazepam, nitrazepam
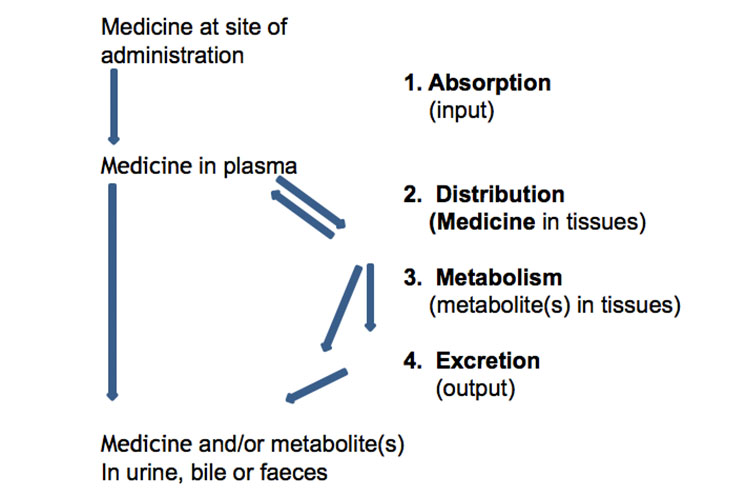
Absorptie, Distributie, Metabolisme en Excretie
Hieronder staat een schematische weergave van de absorptie, distributie, metabolisme en excretie van geneesmiddelen:
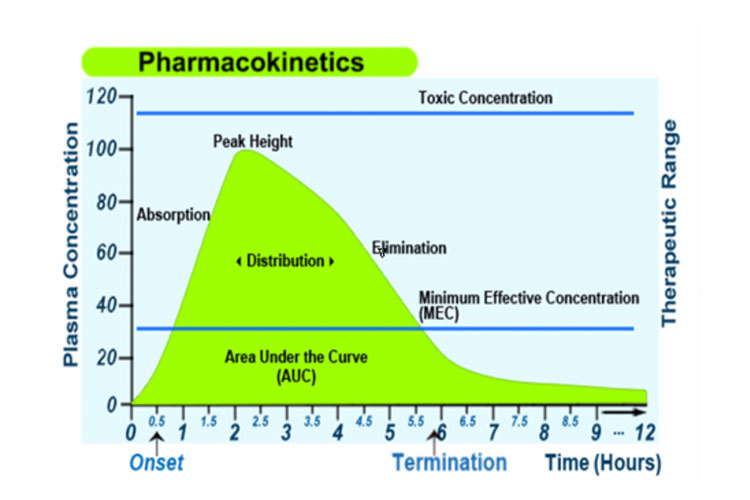
Aternatief geeft de volgende grafiek de absorptie, distributie, metabolisme en uitscheiding van een geneesmiddel weer, samen met enkele farmacokinetische termen, na een enkele orale dosis met onmiddellijke afgifte:
Absorptie
Absorptie vanaf de plaats van toediening maakt het mogelijk dat het therapeutische middel (direct of indirect) in het plasma terechtkomt.
Met het geneesmiddel samenhangende factoren zijn onder meer de ionisatiestatus, het molecuulgewicht, de oplosbaarheid en de formulering. Kleine, niet-geïoniseerde, vetoplosbare geneesmiddelen permeëren de plasmamembranen het gemakkelijkst.
Distributie
Eenmaal geabsorbeerd kan het geneesmiddel vervolgens de bloedbaan reversibel verlaten en zich distribueren in de interstitiële en intracellulaire vloeistoffen.
De permeabiliteit van een geneesmiddel wordt bepaald door de bloed-hersenbarrière, de bloed-testesbarrière en de bloed-placentabarrière.
De meeste geneesmiddelen zijn tot op zekere hoogte eiwitgebonden; alleen een ongebonden geneesmiddel is vrij om zijn farmacologische actie(s) uit te voeren.
Depotopslag heeft betrekking op lipofiele geneesmiddelen die in vet worden opgeslagen, calciumbindende geneesmiddelen, enz.
Met het ouder worden is er een afname van de vetvrije lichaamsmassa en het totale lichaamswatergehalte, en een toename van het totale lichaamsvet. Dit kan leiden tot veranderingen in het distributievolume (Vd) van sommige geneesmiddelen, wat onvoorspelbare effecten kan veroorzaken, met name bij kwetsbare oudere volwassenen.
Het distributievolume is de mate waarin een geneesmiddel zich uit de bloedbaan en in de weefsels van het lichaam verdeelt (d.w.z. de plaats waar de receptoren van het geneesmiddel zich bevinden). Een verlaging van het Vd zal resulteren in hogere plasmaconcentraties voor hydrofiele geneesmiddelen zoals gentamicine, digoxine en lithium. Een hoger percentage lichaamsvet verhoogt de Vd voor lipofiele geneesmiddelen zoals diazepam, waardoor de plasmahalfwaardetijd toeneemt.
Metabolisme
Voordat het geneesmiddel wordt uitgescheiden, wordt het gemetaboliseerd door de lever, de nieren of andere plaatsen. Dit is het proces waarbij het geneesmiddel polairder wordt (beter oplosbaar in water), wat kan leiden tot inactivering en uitscheiding van het geneesmiddel.
Metabolieten kunnen meer of minder (prodrug) actief zijn dan het oorspronkelijke geneesmiddel. De lever is de belangrijkste bron van deze enzymen (P450-enzymen), hoewel ze ook aanwezig kunnen zijn in het maagdarmkanaal, het hart, de longen, de hersenen en de nieren.
1. Fase-I-reacties (niet-synthetisch)
Fase-I-reacties (niet-synthetisch) omvatten kleine structurele wijzigingen van de oorspronkelijke structuur via oxidatie, reductie of hydrolyse om kleinere, beter in water oplosbare metabolieten te produceren. Deze worden voornamelijk afgehandeld door de cytochroom P450-enzymen.
Fase I-reacties bieden vaak een ‘handvat’ voor verdere modificaties door daaropvolgende fase II-reacties.
2. Fase II-reacties (synthetisch)
Fase II-reacties (synthetisch) betreffen de koppeling van een in water oplosbaar endogeen molecuul zoals glucuronzuur, sulfaat of glutathion aan een chemische stof (moederverbinding en/of fase I-metaboliet) om de uitscheiding te vergemakkelijken.
De meest voorkomende oorzaken van interacties tussen geneesmiddelen zijn de farmacokinetiek, met name de metabole interacties. Deze staan bekend als cytochroom P450-interacties.
Een groot aantal klinisch belangrijke interacties ontstaat door remming of inductie van substraten (geneesmiddelen die in belangrijke mate door de gegeven enzymen worden gemetaboliseerd).
- Remmers zijn verbindingen die in het algemeen in staat zijn het metabolisme van de verschillende substraten te remmen. Als gevolg hiervan kan toediening van de remmer leiden tot een verhoogde plasmaconcentratie van het substraat. Zo remt ciprofloxacine het CYP3A4-enzym dat clozapine metaboliseert, wat kan leiden tot een toxiciteit van clozapine.
- Inductoren van de gespecificeerde P450 hebben het vermogen de activiteit van het aangewezen enzym te verhogen en daardoor de plasmaconcentraties van de vermelde substraten te verlagen. Zo induceert carbamazepine het metabolisme van cyclosporine via het CYP3A4-enzym, wat leidt tot een verlaging van de plasmaspiegel van cyclosporine en daarmee tot een verlies van werkzaamheid.

Excretie
Medicijnen en hun metabolieten worden uit het lichaam verwijderd in urine, gal en/of feces. In sommige gevallen moeten ze eerst worden gemetaboliseerd tot beter in water oplosbare verbindingen (voorbeelden zijn amiodaron, amitriptyline, amlodipine, amfotericine B, aripiprazol, aspirine, atomoxetine, atorvastatine, azithromycine, felodipine enz.)
Anderen worden onveranderd of betrekkelijk onveranderd uitgescheiden (penicillines, cefalosporines, aminoglycosiden, ganciclovir, milrinon, oseltamivir, risedronaat, varenicline enz.).
De belangrijkste processen die een rol spelen bij de uitscheiding zijn glomerulaire filtratie, tubulaire secretie en tubulaire reabsorptie.
Tubulaire excretie en reabsorptie kunnen ook worden beïnvloed door geneesmiddelen die de urine zuurder maken (ammoniumchloride, grote doses vitamine C), of juist alkalischer (urine-alkalisatoren zoals Citralite®, Citravescent®, Uracol®, Ural® , die alle de pH-waarde van de urine verhogen tot ongeveer pH 9).
Creatinine en creatinineklaring
Creatinine (normaal bereik: vrouwen 50-110 micromol /L; mannen 60-120 micromol /L) is het stofwisselingsproduct van het spiermetabolisme. Het niveau ervan is een weerspiegeling van zowel de spiermassa als de nierfunctie, aangezien creatinine voornamelijk wordt geëlimineerd door glomerulaire filtratie via de nier.
Lage niveaus kunnen wijzen op eiwithonger, leveraandoeningen of zwangerschap, terwijl hoge niveaus worden waargenomen bij nierfalen, spierdegeneratie en de effecten van sommige geneesmiddelen die de nierafscheiding blokkeren (bijv.
Creatinineklaring (CrCl)
Creatinineklaring (CrCl) is een schatting van de glomerulaire filtratiesnelheid (GFR), die een directe maat is voor de nierfunctie. De CrCl kan worden berekend met behulp van de Cockroft-Gault vergelijking:
(Voor vrouwen, vermenigvuldigen met 0.85 om rekening te houden met de verminderde verhouding tussen spier en ideaal lichaamsgewicht in vergelijking met mannen.)
- Ideaal lichaamsgewicht voor mannen = 50 kg + 0,9 kg/elke cm boven 152 cm.
- Ideaal lichaamsgewicht voor vrouwen = 45,5 kg + 0,9 kg/elke cm boven 152 cm.
(In veel monografieën over geneesmiddelen worden de dosis en de dosisfrequentie van een geneesmiddel aangegeven op basis van de creatinineklaring.)
eGFR
eGFR wordt door pathologielaboratoria gebruikt als een geschatte indicator van de nierfunctie. (eGFR heeft beperkingen, en voor medicijndosering wanneer de nierfunctie <60mL/min is, verdient de Cockcroft-Gault-vergelijking de voorkeur.)
Biobeschikbaarheid
Biobeschikbaarheid is een subcategorie van absorptie en is de fractie van een toegediende dosis onveranderd geneesmiddel die de systemische circulatie bereikt. Wanneer een geneesmiddel intraveneus wordt toegediend, is de (absolute) biologische beschikbaarheid per definitie 100%.
De werkzaamheid van een geneesmiddel is een functie van de snelheid en de mate van absorptie. De relatieve biologische beschikbaarheid wordt afgeleid als de verhouding (%) tussen de biologische beschikbaarheid van een orale dosis (of een andere niet-IV dosis) ten opzichte van een IV dosis.
- Bryant, B & Knights, K 2015, Pharmacology for Health Professionals, 4th edn, Mosby Elsevier, Sydney, Australië
- eMIMs 2016, CMPMedica, https://www.mimsonline.com.au/Login/Login.aspx?ReturnUrl=%2fdefault.aspx
- Harris, P, Nagy, S & Vardaxis, N 2014, Mosby’s Dictionary of Medicine, Nursing and Health Professions, 3rd edn, Elsevier, Australië.
- Neal, M J 2015, Medical Pharmacology at a Glance, 8th edn, Blackwell Science, Oxford.
- Rossi, S 2016, Australian Medicines Handbook (AMH), https://amhonline.amh.net.au/auth