Dopo aver letto e assimilato questo articolo, l’operatore sanitario dovrebbe essere in grado di discutere il significato dei termini farmacocinetica e farmacodinamica; e:
- Confrontare e contrastare la farmacocinetica e la farmacodinamica
- Discutere l’importanza della clearance della creatinina
- Discutere l’importanza dell’emivita di un farmaco
- Discutere l’importanza della via metabolica P450.
Che cos’è la farmacologia
I farmaci mirano a prevenire, curare o controllare vari stati patologici. Per raggiungere questo obiettivo, le concentrazioni adeguate del farmaco devono essere consegnate ai tessuti bersaglio in modo da ottenere livelli terapeutici, ma non tossici.
Le azioni farmacologiche e tossicologiche dei farmaci sono principalmente legate alle loro concentrazioni plasmatiche. Di conseguenza, gli operatori sanitari che lavorano con i farmaci devono riconoscere la velocità di insorgenza dell’azione del farmaco, così come l’intensità e la durata del suo effetto. A loro volta, questi sono controllati dalle seguenti quattro vie fondamentali di movimento e modifica dei farmaci nell’organismo:
- Assorbimento
- Distribuzione
- Metabolismo
- Escrezione, così come l’inizio e la durata dell’azione
Farmacocinetica contro Farmacodinamica
La farmacocinetica influenza la via di somministrazione decisa per un farmaco specifico, la quantità e la frequenza di ogni dose e gli intervalli di dosaggio.
Farmacodinamica, invece, è lo studio di come un farmaco agisce su un organismo vivente. Ciò include la risposta farmacologica e la sua durata e grandezza osservate, relative alla concentrazione del farmaco in un sito attivo nell’organismo; cioè lo studio dell’effetto di un farmaco e dei meccanismi d’azione.
Che cosa fanno i farmaci al corpo
La farmacocinetica clinica è l’applicazione dei principi farmacocinetici e farmacodinamici alla gestione terapeutica sicura ed efficace di un singolo paziente.
Formula dell’emivita
Il termine farmacocinetico emivita (t1/2) si riferisce al tempo impiegato dalla metà della dose iniziale del farmaco somministrato per essere eliminato dal corpo. Cioè:
Per esempio, una dose di farmaco con un’emivita di 12 ore avrebbe il 25% del farmaco rimasto nel corpo dopo 24 ore.
Molti farmaci sono classificati in termini delle loro emivite. Per esempio, le benzodiazepine sono classificate in termini di:
- ad azione ultra-corta (t1/2 < 6 ore): midazolam, triazolam
- ad azione breve (t1/2 6-12 ore): oxazepam, temazepam
- Ad azione media (t1/2 12-24 ore): alprazolam, bromazepam, lorazepam
- Ad azione lunga (t1/2 > 24 ore): clobazepam, clonazepam, diazepam, flunitrazepam, nitrazepam
Assorbimento, distribuzione, metabolismo ed escrezione
Di seguito una rappresentazione schematica dell’assorbimento, distribuzione, metabolismo ed escrezione dei farmaci:
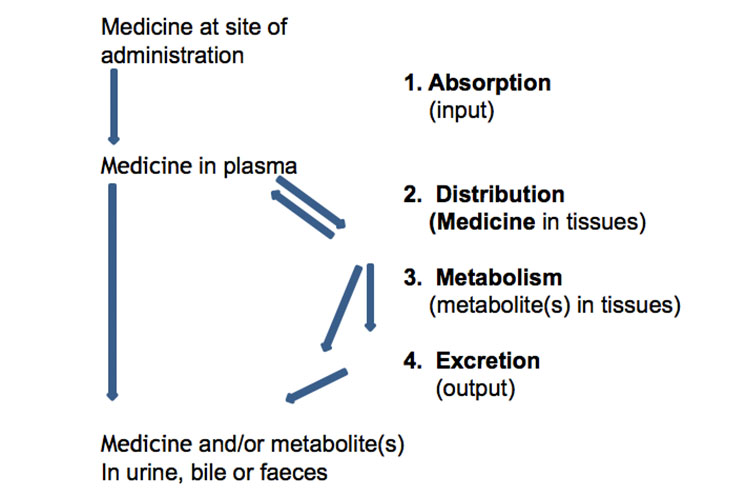
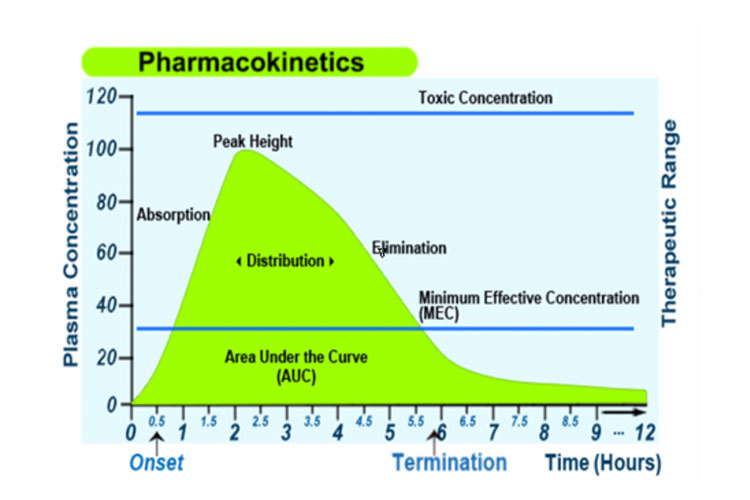
In alternativa, il seguente grafico rappresenta l’assorbimento, la distribuzione, il metabolismo e l’escrezione di un farmaco, insieme ad alcuni termini farmacocinetici, dopo una singola dose orale a rilascio immediato:
Assorbimento
L’assorbimento dal sito di somministrazione permette l’ingresso dell’agente terapeutico (direttamente o indirettamente) nel plasma.
I fattori legati alla medicina includono lo stato di ionizzazione, il peso molecolare, la solubilità e la formulazione. I farmaci piccoli, non ionizzati e solubili nei lipidi permeano più facilmente le membrane plasmatiche.
Distribuzione
Una volta assorbito, il farmaco può quindi lasciare reversibilmente il flusso sanguigno e distribuirsi nei fluidi interstiziali e intracellulari.
La permeabilità di un farmaco è definita dalla barriera emato-encefalica, dalla barriera emato-testuale e dalla barriera emato-placentare.
La maggior parte dei farmaci sono in qualche misura legati alle proteine; solo un farmaco non legato è libero di svolgere la sua azione farmacologica.
Il deposito si riferisce ai farmaci lipofili che si conservano nel grasso, ai farmaci che legano il calcio, ecc.
Con l’invecchiamento, si verifica una riduzione della massa magra e del contenuto totale di acqua corporea, e un aumento del grasso corporeo totale. Questo può portare a cambiamenti nel volume di distribuzione (Vd) per alcuni farmaci, causando effetti imprevedibili, in particolare negli adulti anziani fragili.
Il volume di distribuzione è la misura in cui un farmaco si distribuisce fuori dal flusso sanguigno e nei tessuti del corpo (cioè il sito dei recettori del farmaco). Una diminuzione del Vd risulterà in concentrazioni plasmatiche più alte per i farmaci idrofili come la gentamicina, la digossina e il litio. Una maggiore proporzione di grasso corporeo aumenterà la Vd per i farmaci lipofili come il diazepam, causando un aumento dell’emivita plasmatica.
Metabolismo
Prima di essere escreto, il farmaco viene metabolizzato da fegato, reni o altri siti. Questo è il processo che rende il farmaco più polare (più solubile in acqua), che può portare all’inattivazione del farmaco e all’escrezione.
I metaboliti possono essere più o meno (prodrug) attivi del farmaco padre. Il fegato è la fonte principale di questi enzimi (enzimi P450), anche se possono essere presenti nel tratto gastrointestinale, cuore, polmoni, cervello e reni.
1. Reazioni di fase I (non sintetiche)
Le reazioni di fase I (non sintetiche) comportano piccole modifiche strutturali della struttura madre tramite ossidazione, riduzione o idrolisi per produrre metaboliti più piccoli e solubili in acqua. Queste sono gestite principalmente dagli enzimi del citocromo P450.
Le reazioni di fase I spesso forniscono un “manico” per ulteriori modifiche da parte delle successive reazioni di fase II.
2. Reazioni di fase II (sintetiche)
Le reazioni di fase II (sintetiche) comportano l’accoppiamento di una molecola endogena idrosolubile come l’acido glucuronico, il solfato o il glutatione a una sostanza chimica (composto madre e/o metabolita di fase I) per facilitare l’escrezione.
Le cause più comuni delle interazioni medicinale-medicina sono la farmacocinetica, in particolare quella metabolica. Queste sono note come interazioni del citocromo P450.
Un gran numero di interazioni clinicamente importanti derivano dall’inibizione o dall’induzione dei substrati (farmaci che sono significativamente metabolizzati da determinati enzimi).
- Gli inibitori sono composti che sono generalmente capaci di inibire il metabolismo dei vari substrati. Di conseguenza, la somministrazione dell’inibitore può portare ad un aumento della concentrazione plasmatica del substrato. Per esempio, la ciprofloxacina inibisce l’enzima CYP3A4 che metabolizza la clozapina, il che può portare a una tossicità della clozapina.
- Gli induttori del P450 specificato hanno la capacità di aumentare l’attività dell’enzima designato e quindi ridurre le concentrazioni plasmatiche dei substrati elencati. Per esempio, la carbamazepina induce il metabolismo della ciclosporina attraverso l’enzima CYP3A4, portando ad una riduzione dei livelli plasmatici della ciclosporina e quindi alla perdita di efficacia.
 iv I farmaci e il corpo
iv I farmaci e il corpoEscrezione
I farmaci e i loro metaboliti sono rimossi dal corpo nelle urine, bile e/o feci. In alcuni casi, devono prima essere metabolizzati in società più solubili in acqua (esempi: amiodarone, amitriptilina, amlodipina, amfotericina B, aripiprazolo, aspirina, atomoxetina, atorvastatina, azitromicina, felodipina ecc.)
Altri vengono escreti invariati o relativamente invariati (penicilline, cefalosporine, aminoglicosidi, ganciclovir, milrinone, oseltamivir, risedronato, vareniclina ecc.
L’escrezione e il riassorbimento tubulare possono anche essere influenzati da farmaci che rendono l’urina più acida (cloruro di ammonio, grandi dosi di vitamina C), o più alcalina (alcalinizzanti urinari come Citralite®, Citravescent®, Uracol®, Ural® , che alzano il pH urinario a circa pH 9).
Creatinina e clearance della creatinina
La creatinina (range di normalità: femmine 50-110 micromol /L; maschi 60-120 micromol /L) è il prodotto metabolico del metabolismo muscolare. Il suo livello è un riflesso sia della massa muscolare che della funzione renale, in quanto la creatinina viene eliminata prevalentemente per filtrazione glomerulare attraverso il rene.
Livelli bassi possono indicare fame di proteine, malattie epatiche o gravidanza, mentre livelli elevati si riscontrano nell’insufficienza renale, nella degenerazione muscolare e negli effetti di alcuni farmaci che bloccano la secrezione renale (es.
Creatinina clearance (CrCl)
La clearance della creatinina (CrCl) è una stima della velocità di filtrazione glomerulare (GFR), che è una misura diretta della funzione renale. La CrCl può essere calcolata per mezzo dell’equazione di Cockroft-Gault:
(Per le donne, moltiplicare per 0.85 per tenere conto del ridotto rapporto tra muscoli e peso corporeo ideale rispetto ai maschi.)
- Peso corporeo ideale per i maschi = 50 kg + 0,9 kg/ogni cm sopra i 152 cm.
- Peso corporeo ideale per le femmine = 45,5 kg + 0,9 kg/ogni cm sopra i 152 cm.
(Molte monografie sui farmaci indicano la dose e la frequenza di dosaggio di un farmaco in base alla clearance della creatinina.)
eGFR
eGFR è usato dai laboratori di patologia come un indicatore stimato della funzione renale. (L’eGFR ha dei limiti, e per il dosaggio dei farmaci quando la funzione renale è <60mL/min, l’equazione di Cockcroft-Gault è preferibile.)
Biodisponibilità
La biodisponibilità è una sottocategoria dell’assorbimento ed è la frazione di una dose somministrata di farmaco invariato che raggiunge la circolazione sistemica. Per definizione, quando un farmaco viene somministrato per via endovenosa, la sua biodisponibilità (assoluta) è del 100%.
L’efficacia di un farmaco è una funzione del tasso e dell’estensione dell’assorbimento. La biodisponibilità relativa è derivata come un rapporto (%) tra la biodisponibilità da una forma di dose orale (o altra dose non-IV) rispetto a una dose IV.
- Bryant, B & Knights, K 2015, Pharmacology for Health Professionals, 4th edn, Mosby Elsevier, Sydney, Australia
- eMIMs 2016, CMPMedica, https://www.mimsonline.com.au/Login/Login.aspx?ReturnUrl=%2fdefault.aspx
- Harris, P, Nagy, S & Vardaxis, N 2014, Mosby’s Dictionary of Medicine, Nursing and Health Professions, 3rd edn, Elsevier, Australia.
- Neal, M J 2015, Medical Pharmacology at a Glance, 8th edn, Blackwell Science, Oxford.
- Rossi, S 2016, Australian Medicines Handbook (AMH), https://amhonline.amh.net.au/auth