Après avoir lu et digéré cet article, le professionnel de santé en exercice devrait être en mesure de discuter de la signification des termes pharmacocinétique et pharmacodynamique ; et :
- Comparer et opposer pharmacocinétique et pharmacodynamique
- Discuter de l’importance de la clairance de la créatinine
- Discuter de l’importance de la demi-vie d’un médicament
- Discuter de l’importance de la voie métabolique P450.
Qu’est-ce que la pharmacologie
Les médicaments visent à prévenir, guérir ou contrôler divers états pathologiques. Pour atteindre cet objectif, des concentrations adéquates du médicament doivent être délivrées aux tissus cibles de manière à obtenir des niveaux thérapeutiques, mais non toxiques.
Les actions pharmacologiques et toxicologiques des médicaments sont principalement liées à leurs concentrations plasmatiques. Par conséquent, les professionnels de santé qui travaillent avec des médicaments doivent reconnaître la vitesse d’apparition de l’action du médicament ainsi que l’intensité et la durée de son effet. À leur tour, celles-ci sont contrôlées par les quatre voies fondamentales suivantes de déplacement et de modification des médicaments dans l’organisme :
- Absorption
- Distribution
- Métabolisme
- Excrétion, ainsi que le début et la durée d’action
Pharmacocinétique v Pharmacodynamique
La pharmacocinétique influence la voie d’administration décidée pour un médicament spécifique, la quantité et la fréquence de chaque dose et ses intervalles d’administration.
La pharmacodynamique, quant à elle, est l’étude de la façon dont un médicament agit sur un organisme vivant . Cela comprend la réponse pharmacologique, sa durée et son ampleur observées, par rapport à la concentration du médicament sur un site actif de l’organisme ; c’est-à-dire l’étude de l’effet d’un médicament et des mécanismes d’action.
Ce que les médicaments font au corps
La pharmacocinétique clinique est l’application des principes pharmacocinétiques et pharmacodynamiques à la prise en charge thérapeutique sûre et efficace d’un patient individuel.
Formule de demi-vie
Le terme pharmacocinétique demi-vie (t1/2) désigne le temps nécessaire pour que la moitié de la dose initiale de médicament administrée soit éliminée de l’organisme. C’est-à-dire :
Par exemple, une dose de médicament ayant une demi-vie de 12 heures aurait 25% du médicament restant dans le corps après 24 heures.
De nombreux médicaments sont classés en fonction de leur demi-vie. Par exemple, les benzodiazépines sont classées en termes de :
- Action ultra-courte (t1/2 < 6 heures) : midazolam, triazolam
- Action courte (t1/2 6-12 heures) : oxazépam, témazépam
- Action moyenne (t1/2 12-24 heures) : alprazolam, bromazépam, lorazépam
- Action longue (t1/2 > 24 heures) : clobazépam, clonazépam, diazépam, flunitrazépam, nitrazépam
Absorption, distribution, métabolisme et excrétion
Vous trouverez ci-dessous une représentation schématique de l’absorption, de la distribution, du métabolisme et de l’excrétion des médicaments :
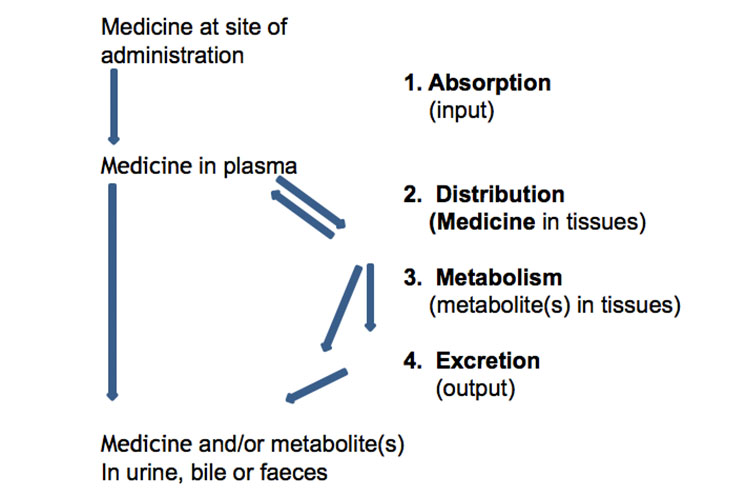
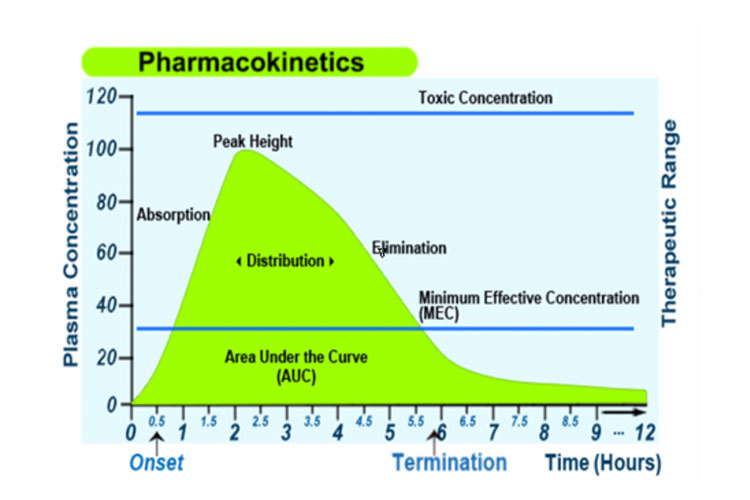
Alternativement, le graphique suivant représente l’absorption, la distribution, le métabolisme et l’excrétion d’un médicament, ainsi que certains termes pharmacocinétiques, après une dose unique orale à libération immédiate :
Absorption
L’absorption à partir du site d’administration permet l’entrée de l’agent thérapeutique (directement ou indirectement) dans le plasma.
Les facteurs liés à la médecine comprennent l’état d’ionisation, le poids moléculaire, la solubilité et la formulation. Les médicaments de petite taille, non ionisés et liposolubles, traversent le plus facilement les membranes plasmatiques.
Distribution
Une fois absorbé, le médicament peut alors quitter de manière réversible la circulation sanguine et se distribuer dans les liquides interstitiels et intracellulaires.
La perméabilité d’un médicament est définie par la barrière hémato-encéphalique, la barrière hémato-testiculaire et la barrière sang-placenta.
La plupart des médicaments sont liés aux protéines dans une certaine mesure ; seul un médicament non lié est libre d’exercer sa ou ses actions pharmacologiques.
La conservation au dépôt concerne les médicaments lipophiles qui se stockent dans les graisses, les médicaments liant le calcium, etc.
Avec le vieillissement, on observe une réduction de la masse maigre et de la teneur totale en eau du corps, et une augmentation de la masse grasse totale. Cela peut entraîner des modifications du volume de distribution (Vd) de certains médicaments, provoquant des effets imprévisibles, notamment chez les personnes âgées fragiles.
Le volume de distribution est la mesure dans laquelle un médicament se distribue hors de la circulation sanguine et dans les tissus de l’organisme (c’est-à-dire le site des récepteurs du médicament). Une diminution du Vd entraînera des concentrations plasmatiques plus élevées pour les médicaments hydrophiles tels que la gentamicine, la digoxine et le lithium. Une proportion plus élevée de graisse corporelle augmentera la Vd pour les médicaments lipophiles tels que le diazépam, entraînant une augmentation de la demi-vie plasmatique.
Métabolisme
Avant d’être excrété, le médicament est métabolisé par le foie, les reins ou d’autres sites. Ce processus consiste à rendre le médicament plus polaire (plus soluble dans l’eau), ce qui peut conduire à l’inactivation du médicament et à son excrétion.
Les métabolites peuvent être plus ou moins actifs (prodrogues) que le médicament parent. Le foie est la principale source de ces enzymes (enzymes P450), bien qu’ils puissent être présents dans le tractus gastro-intestinal, le cœur, les poumons, le cerveau et les reins.
1. Réactions de phase I (non synthétiques)
Les réactions de phase I (non synthétiques) impliquent des modifications structurelles mineures de la structure parentale par oxydation, réduction ou hydrolyse pour produire des métabolites plus petits et plus hydrosolubles. Ceux-ci sont principalement pris en charge par les enzymes du cytochrome P450.
Les réactions de phase I fournissent fréquemment une » poignée » pour d’autres modifications par des réactions de phase II ultérieures.
2. Réactions de phase II (synthétiques)
Les réactions de phase II (synthétiques) impliquent le couplage d’une molécule endogène hydrosoluble telle que l’acide glucuronique, le sulfate ou le glutathion à un produit chimique (composé parent et/ou métabolite de phase I) afin de faciliter l’excrétion.
Les causes les plus courantes d’interactions entre médicaments sont la pharmacocinétique, en particulier métabolique. Elles sont connues sous le nom d’interactions du cytochrome P450.
Un grand nombre d’interactions cliniquement importantes résultent de l’inhibition ou de l’induction de substrats (médicaments qui sont significativement métabolisés par les enzymes données).
- Les inhibiteurs sont des composés qui sont généralement capables d’inhiber le métabolisme des différents substrats. Par conséquent, l’administration de l’inhibiteur peut entraîner une augmentation de la concentration plasmatique du substrat. Par exemple, la ciprofloxacine inhibe l’enzyme CYP3A4 qui métabolise la clozapine, ce qui peut conduire à une toxicité de la clozapine.
- Les inducteurs du P450 spécifié ont la capacité d’augmenter l’activité de l’enzyme désignée et donc de réduire les concentrations plasmatiques des substrats listés. Par exemple, la carbamazépine induit le métabolisme de la ciclosporine via l’enzyme CYP3A4, entraînant une réduction des concentrations plasmatiques de la ciclosporine et donc une perte d’efficacité.
 . Les médicaments et le corps
. Les médicaments et le corpsExcrétion
Les médicaments et leurs métabolites sont éliminés du corps par l’urine, la bile et/ou les fèces. Dans certains cas, ils doivent d’abord être métabolisés en fractions plus hydrosolubles (par exemple, l’amiodarone, l’amitriptyline, l’amlodipine, l’amphotéricine B, l’aripiprazole, l’aspirine, l’atomoxétine, l’atorvastatine, l’azithromycine, la félodipine, etc.)
D’autres sont excrétés sous forme inchangée ou relativement inchangée (pénicillines, céphalosporines, aminoglycosides, ganciclovir, milrinone, oseltamivir, risédronate, varénicline, etc.).
Les principaux processus impliqués dans l’excrétion sont la filtration glomérulaire, la sécrétion tubulaire et la réabsorption tubulaire.
L’excrétion et la réabsorption tubulaires peuvent également être influencées par des médicaments qui rendent l’urine soit plus acide (chlorure d’ammonium, fortes doses de vitamine C), soit plus alcaline (alcalinisants urinaires tels que Citralite®, Citravescent®, Uracol®, Ural®, qui élèvent tous le pH urinaire à environ pH 9).
Créatinine et clairance de la créatinine
La créatinine (fourchette normale : femmes 50-110 micromol /L ; hommes 60-120 micromol /L) est le produit du métabolisme musculaire. Son taux est le reflet à la fois de la masse musculaire et de la fonction rénale, car la créatinine est principalement éliminée par filtration glomérulaire à travers le rein.
Des taux faibles peuvent indiquer une privation de protéines, une maladie du foie ou une grossesse, tandis que des taux élevés sont observés en cas d’insuffisance rénale, de dégénérescence musculaire et des effets de certains médicaments qui bloquent la sécrétion rénale (par ex.par exemple la cimétidine et le triméthoprime).
La clairance de la créatinine (CrCl)
La clairance de la créatinine (CrCl) est une estimation du débit de filtration glomérulaire (DFG), qui est une mesure directe de la fonction rénale. La ClCr peut être calculée au moyen de l’équation de Cockroft-Gault :
(Pour les femmes, multiplier par 0.85 pour tenir compte de la réduction du rapport muscle/poids corporel idéal par rapport aux hommes.)
- Poids corporel idéal pour les hommes = 50 kg + 0,9 kg/chaque cm au-dessus de 152 cm.
- Poids corporel idéal pour les femmes = 45,5 kg + 0,9 kg/chaque cm au-dessus de 152 cm.
(De nombreuses monographies de médicaments indiqueront la dose et la fréquence des doses d’un médicament en fonction de la clairance de la créatinine.)
Le DFGe
Le DFGe est utilisé par les laboratoires de pathologie comme indicateur estimé de la fonction rénale. (Le DFGe a des limites, et pour le dosage des médicaments lorsque la fonction rénale est < 60mL/min, l’équation de Cockcroft-Gault est préférable.)
Biodisponibilité
La biodisponibilité est une sous-catégorie de l’absorption et représente la fraction d’une dose administrée de médicament inchangé qui atteint la circulation systémique. Par définition, lorsqu’un médicament est administré par voie intraveineuse, sa biodisponibilité (absolue) est de 100 %.
L’efficacité d’un médicament est fonction de la vitesse et de l’étendue de l’absorption. La biodisponibilité relative est dérivée comme une relation (%) entre la biodisponibilité d’une forme posologique orale (ou autre dose non IV) par rapport à une dose IV.
- Bryant, B & Knights, K 2015, Pharmacology for Health Professionals, 4th edn, Mosby Elsevier, Sydney, Australia
- eMIMs 2016, CMPMedica, https://www.mimsonline.com.au/Login/Login.aspx?ReturnUrl=%2fdefault.aspx
- Neal, M J 2015, Medical Pharmacology at a Glance, 8th edn, Blackwell Science, Oxford.
- Rossi, S 2016, Australian Medicines Handbook (AMH), https://amhonline.amh.net.au/auth
Harris, P, Nagy, S & Vardaxis, N 2014, Mosby’s Dictionary of Medicine, Nursing and Health Professions, 3e éd., Elsevier, Australie.
.