Después de leer y digerir este artículo, el profesional sanitario en activo debe ser capaz de discutir el significado de los términos farmacocinética y farmacodinámica; y:
- Comparar y contrastar la farmacocinética y la farmacodinámica
- Discutir la importancia del aclaramiento de creatinina
- Discutir la importancia de la vida media de un medicamento
- Discutir la importancia de la vía metabólica P450.
Qué es la Farmacología
Los medicamentos tienen como objetivo prevenir, curar o controlar diversos estados de enfermedad. Para lograr este objetivo, se deben suministrar concentraciones adecuadas del medicamento a los tejidos diana, de manera que se obtengan niveles terapéuticos, pero no tóxicos.
Las acciones farmacológicas y toxicológicas de los medicamentos están relacionadas principalmente con sus concentraciones plasmáticas. En consecuencia, los profesionales sanitarios que trabajan con medicamentos deben reconocer la velocidad de inicio de la acción del medicamento, así como la intensidad y duración de su efecto. A su vez, estos están controlados por las siguientes cuatro vías fundamentales de movimiento y modificación de los medicamentos en el organismo:
- Absorción
- Distribución
- Metabolismo
- Excreción, así como el inicio y la duración de la acción
Farmacocinética v Farmacodinámica
La farmacocinética influye en la vía de administración decidida para un medicamento específico, la cantidad y la frecuencia de cada dosis y sus intervalos de dosificación.
La farmacodinámica, en cambio, es el estudio de cómo actúa un medicamento en un organismo vivo . Esto incluye la respuesta farmacológica y su duración y magnitud observadas, en relación con la concentración del medicamento en un sitio activo del organismo; es decir, el estudio del efecto de un medicamento y los mecanismos de acción.
Qué hacen los medicamentos en el organismo
La farmacocinética clínica es la aplicación de los principios farmacocinéticos y farmacodinámicos al manejo terapéutico seguro y eficaz de un paciente individual.
Fórmula de la vida media
El término farmacocinético vida media (t1/2) se refiere al tiempo que tarda en eliminarse del organismo la mitad de la dosis inicial del medicamento administrado. Es decir:
Por ejemplo, una dosis medicamentosa con una vida media de 12 horas tendría un 25% del medicamento restante en el cuerpo después de 24 horas.
Muchos medicamentos se clasifican en términos de su vida media. Por ejemplo, las benzodiacepinas se clasifican en términos de:
- De acción ultracorta (t1/2 < 6 horas): midazolam, triazolam
- De acción corta (t1/2 6-12 horas): oxazepam, temazepam
- De acción media (t1/2 12-24 horas): alprazolam, bromazepam, lorazepam
- De acción larga (t1/2 > 24 horas): clobazepam, clonazepam, diazepam, flunitrazepam, nitrazepam
Absorción, distribución, metabolismo y excreción
A continuación se muestra una representación esquemática de la absorción, distribución, metabolismo y excreción de los medicamentos:
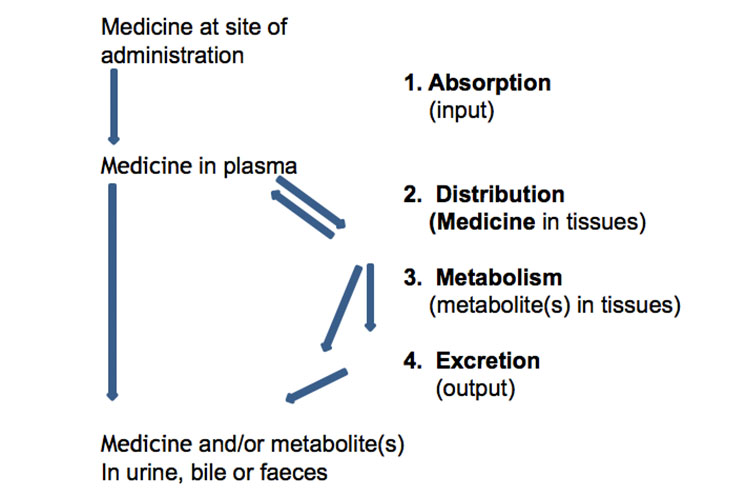
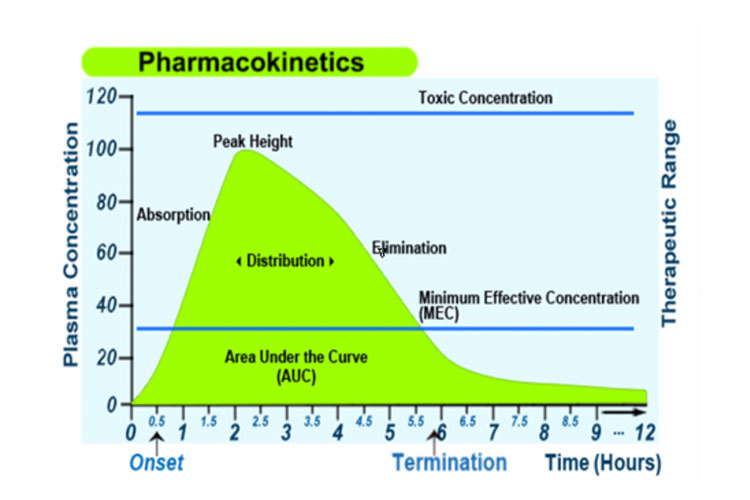
Alternativamente, el siguiente gráfico representa la absorción, distribución, metabolismo y excreción de un medicamento, junto con algunos términos farmacocinéticos, tras una única dosis oral de liberación inmediata:
Absorción
La absorción desde el lugar de administración permite la entrada del agente terapéutico (directa o indirectamente) en el plasma.
Los factores relacionados con el medicamento incluyen el estado de ionización, el peso molecular, la solubilidad y la formulación. Los medicamentos pequeños, no ionizados y solubles en lípidos son los que más fácilmente permean las membranas plasmáticas.
Distribución
Una vez absorbido, el medicamento puede salir reversiblemente del torrente sanguíneo y distribuirse en los fluidos intersticiales e intracelulares.
La permeabilidad de un medicamento viene definida por la barrera hematoencefálica, la barrera hemato-testicular y la barrera hemato-placentaria.
La mayoría de los medicamentos están unidos a proteínas en cierta medida; sólo un medicamento no unido es libre de llevar a cabo su(s) acción(es) farmacológica(s).
El almacenamiento en depósito se refiere a los medicamentos lipofílicos que se almacenan en la grasa, los fármacos que se unen al calcio, etc.
Con el envejecimiento, se produce una reducción de la masa corporal magra y del contenido total de agua corporal, y un aumento de la grasa corporal total. Esto puede dar lugar a cambios en el volumen de distribución (Vd) de algunos medicamentos, causando efectos imprevisibles, especialmente en adultos mayores frágiles.
El volumen de distribución es la medida en que un medicamento se distribuye fuera del torrente sanguíneo y en los tejidos del cuerpo (es decir, el lugar de los receptores del fármaco). Una disminución del Vd dará lugar a concentraciones plasmáticas más elevadas para los medicamentos hidrofílicos como la gentamicina, la digoxina y el litio. Una mayor proporción de grasa corporal aumentará el Vd para los medicamentos lipofílicos como el diazepam, provocando un aumento de la semivida plasmática.
Metabolismo
Antes de ser excretado, el medicamento es metabolizado por el hígado, el riñón u otros sitios. Este proceso hace que el medicamento sea más polar (más soluble en agua), lo que puede conducir a la inactivación del medicamento y a su excreción.
Los metabolitos pueden ser más o menos (profármacos) activos que el medicamento original. El hígado es la principal fuente de estas enzimas (enzimas P450), aunque pueden estar presentes en el tracto gastrointestinal, el corazón, los pulmones, el cerebro y los riñones.
1. Reacciones de fase I (no sintéticas)
Las reacciones de fase I (no sintéticas) implican pequeñas modificaciones estructurales de la estructura madre mediante oxidación, reducción o hidrólisis para producir metabolitos más pequeños y solubles en agua. Éstas son manejadas predominantemente por las enzimas del citocromo P450.
Las reacciones de fase I suelen proporcionar un «asidero» para modificaciones posteriores mediante reacciones de fase II.
2. Reacciones de fase II (sintéticas) Reacciones de fase II (sintéticas)
Las reacciones de fase II (sintéticas) implican el acoplamiento de una molécula endógena hidrosoluble, como el ácido glucurónico, el sulfato o el glutatión, a una sustancia química (compuesto madre y/o metabolito de fase I) para facilitar su excreción.
Las causas más comunes de las interacciones entre medicamentos son las farmacocinéticas, especialmente las metabólicas. Se conocen como interacciones del citocromo P450.
Un gran número de interacciones clínicamente importantes surgen de la inhibición o inducción de los sustratos (medicamentos que se metabolizan significativamente por las enzimas dadas).
- Los inhibidores son compuestos que generalmente son capaces de inhibir el metabolismo de los distintos sustratos. Como resultado, la administración del inhibidor puede conducir a un aumento de la concentración plasmática del sustrato. Por ejemplo, la ciprofloxacina inhibe la enzima CYP3A4 que metaboliza la clozapina, lo que puede dar lugar a una toxicidad de la clozapina.
- Los inductores del P450 especificado tienen la capacidad de aumentar la actividad de la enzima designada y, por tanto, de reducir las concentraciones plasmáticas de los sustratos enumerados. Por ejemplo, la carbamazepina induce el metabolismo de la ciclosporina a través de la enzima CYP3A4, lo que conduce a una reducción de los niveles plasmáticos de la ciclosporina y, por tanto, a la pérdida de eficacia.

Excreción
Los medicamentos y sus metabolitos se eliminan del cuerpo en la orina, la bilis y/o las heces. En algunos casos, primero tienen que ser metabolizados a partes más hidrosolubles (ejemplos: amiodarona, amitriptilina, amlodipino, anfotericina B, aripiprazol, aspirina, atomoxetina, atorvastatina, azitromicina, felodipino, etc.).)
Otros se excretan sin cambios o relativamente sin cambios (penicilinas, cefalosporinas, aminoglucósidos, ganciclovir, milrinona, oseltamivir, risedronato, vareniclina, etc.).
Los principales procesos implicados en la excreción son la filtración glomerular, la secreción tubular y la reabsorción tubular.
La excreción y la reabsorción tubular también pueden verse influenciadas por medicamentos que hacen que la orina sea más ácida (cloruro de amonio, grandes dosis de vitamina C), o más alcalina (alcalinizantes urinarios como Citralite®, Citravescent®, Uracol®, Ural® , que elevan el pH urinario a aproximadamente pH 9).
Creatinina y aclaramiento de creatinina
La creatinina (rango normal: mujeres 50-110 micromol /L; hombres 60-120 micromol /L) es el producto metabólico del metabolismo muscular. Su nivel es un reflejo tanto de la masa muscular como de la función renal, ya que la creatinina se elimina predominantemente por filtración glomerular a través del riñón.
Los niveles bajos pueden indicar inanición proteica, enfermedad hepática o embarazo, mientras que los niveles altos se observan en la insuficiencia renal, en la degeneración muscular y en los efectos de algunos medicamentos que bloquean la secreción renal (p.Por ejemplo, la cimetidina y la trimetoprima).
Aclaramiento de creatinina (CrCl)
El aclaramiento de creatinina (CrCl) es una estimación de la tasa de filtración glomerular (TFG), que es una medida directa de la función renal. El CrCl puede calcularse mediante la ecuación de Cockroft-Gault:
(Para las mujeres, multiplicar por 0.85 para tener en cuenta la reducción de la relación entre el músculo y el peso corporal ideal en comparación con los hombres.)
- Peso corporal ideal para hombres = 50 kg + 0,9 kg/cada cm por encima de 152 cm.
- Peso corporal ideal para mujeres = 45,5 kg + 0,9 kg/cada cm por encima de 152 cm.
(Muchas monografías sobre medicamentos indicarán la dosis y la frecuencia de la dosis de un medicamento según el aclaramiento de creatinina.)
FGe
El FGe es utilizado por los laboratorios de patología como un indicador estimado de la función renal. (La TFGe tiene limitaciones, y para la dosificación de medicamentos cuando la función renal es <60mL/min, es preferible la ecuación de Cockcroft-Gault.)
Disponibilidad biológica
La biodisponibilidad es una subcategoría de la absorción y es la fracción de una dosis administrada de un medicamento inalterado que llega a la circulación sistémica. Por definición, cuando un medicamento se administra por vía intravenosa, su biodisponibilidad (absoluta) es del 100%.
La eficacia de un medicamento es una función de la tasa y el grado de absorción. La biodisponibilidad relativa se obtiene como una relación (%) entre la biodisponibilidad de una dosis oral (u otra dosis no intravenosa) en relación con una dosis intravenosa.
.