Estos son los apuntes de la clase 11 del curso de Biología Celular de Harvard Extension.
Esta clase cubrirá dos formas diferentes en las que las células pueden morir: apoptosis (muerte celular programada) y necrosis (muerte celular no planificada). Es fácil diferenciarlas morfológicamente bajo el microscopio, como se muestra en esta imagen de Wikimedia Commons:
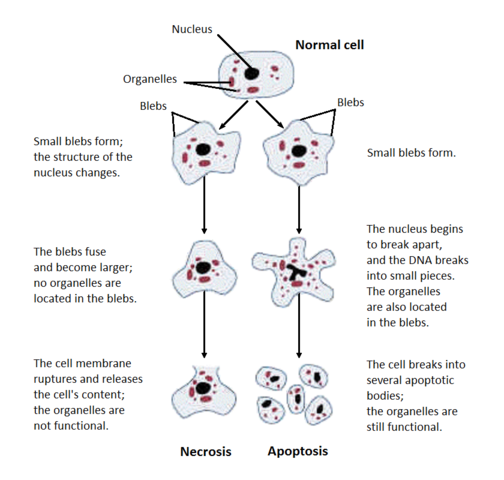
La necrosis es cuando las células mueren accidentalmente debido, por ejemplo, a un traumatismo (por ejemplo, la picadura de una araña venenosa), o a la falta de nutrientes (por ejemplo, la falta de suministro de sangre). La necrosis comienza con la hinchazón de la célula, la cromatina se digiere, las membranas plasmáticas y de los orgánulos se rompen, el RE se vacuoliza, los orgánulos se rompen por completo y, finalmente, la célula se lisa, arrojando su contenido intracelular y provocando una respuesta inmune (inflamación).
La apoptosis puede constituir un suicidio o asesinato celular. Las células se suicidan cuando carecen de cualquier señal de supervivencia en forma de factores tróficos, o cuando detectan daños extensos en el ADN de su propio núcleo. Las células asesinarán a otras células para eliminar las células innecesarias o para eliminar las células inmunitarias potencialmente autoagresivas.
Cualquiera de estos procesos constituye una muerte celular programada. Durante el desarrollo embrionario, las personas tienen manos y pies palmeados y cola; las células que constituyen esas partes se apoptizan posteriormente. La apoptosis también se produce constantemente en muchos tejidos, incluidos los intestinos.
Aquí tienes una impresionante imagen de Wikimedia Commons sobre la apoptosis (léase de izquierda a derecha, de arriba a abajo) gracias a Egelberg:
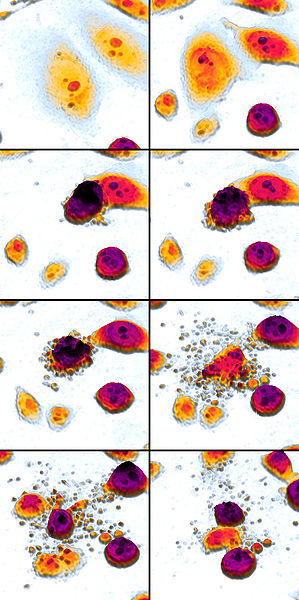
Pasos principales de la apoptosis:
- La célula se encoge
- La célula se fragmenta
- El citoesqueleto se colapsa
- La envoltura nuclear se desensambla
- Las células liberan cuerpos apoptóticos
- ‘proteínas asesinas’: las caspasas (se discuten en detalle más adelante).
- Proteínas de destrucción’ que digieren el ADN, fragmentan la célula y rompen el citoesqueleto
- Proteínas de engullimiento’ que provocan y promueven la fagocitosis por parte de otras células
- CED-3 aprieta el gatillo, activando las proteínas apoptóticas que destruyen la célula. (En el equivalente de los mamíferos, la CED-3 es la Caspasa 9, que escinde y activa la Caspasa 3, que a su vez destruye la célula)
- La CED-4 activa la CED-3.
- CED-9 se une a CED-4, impidiendo su activación
- EGL-1 se activa transcripcionalmente en respuesta a las señales de muerte y cataliza la liberación de CED-4 de CED-9.
Notablemente, en esta lista está ausente ‘enviar una señal.’ Las células apoptóticas no envían ninguna señal, con una excepción: liberan cuerpos apoptóticos y ‘proteínas de engullimiento’ para inducir a otras células (células ‘fagocíticas’) a engullir los cuerpos apoptóticos y y descomponerlos en sus lisosomas, pero esto no es una gran respuesta inmune.
Proteínas importantes en la apoptosis:
C. elegans ha sido el principal organismo modelo para entender la apoptosis, tanto por genética directa como inversa. La genética directa consiste en observar un fenotipo y luego determinar qué gen lo origina; la genética inversa consiste en introducir una mutación en un gen conocido para ver qué fenotipo resulta.
La vía clave en la apoptosis de C. elegans se muestra en este dibujo de Google que he creado:
Aquí hay una explicación de cómo cada una de estas proteínas hace su trabajo, de abajo hacia arriba:
son:
Nótese que no hay robustez en este sistema – se trata de puntos únicos de fallo hasta el final. Si se elimina CED-3, no puede producirse apoptosis. Si se elimina CED-4, no puede producirse apoptosis. Si se elimina el CED-9, todas las células del gusano se apoptarán. Si se elimina EGL-1, no puede producirse apoptosis. Nótese que el orden en que apuntan las flechas en el diagrama anterior refleja el flujo de información en el sistema. Por ejemplo, si se eliminan EGL-1 y CED-9, ocurre lo mismo que si se elimina sólo CED-9: todas las células se apoptan.
En los mamíferos, la apoptosis se rige principalmente por las caspasas (proteasas cisteína-aspárticas). Toda la vía de las caspasas está regulada postraduccionalmente: las caspasas están siempre presentes en forma inactiva (denominadas procaspasas, que contienen un subdominio, el cual contiene un dominio de reclutamiento de caspasas (CARD)) y pueden ser activadas por escisión. Esto permite una respuesta muy rápida si se necesita el suicidio celular. Para que se produzca la apoptosis, las caspasas iniciadoras deben escindirse y dimerizarse. Una vez activadas, deben escindir las caspasas efectoras (también conocidas como pro-caspasas), desencadenando una «cascada de caspasas». Esto amplifica el número de caspasas activadas en la célula. Las caspasas efectoras tienen muchos objetivos, incluyendo la lámina nuclear y el citoesqueleto.
Existen caspasas pro-supervivencia y pro-apoptóticas, y comparten muchos dominios comunes. Las caspasas pro-supervivencia tienen BH1, 2, 3 y 4; las caspasas pro-apoptosis tienen BH1, 2 y 3 o sólo BH3.
Las proteínas inhibidoras de la apoptosis (IAPs) frenan tanto las caspasas iniciadoras como las efectoras. Cada una de ellas tiene un dominio de unión de zinc que se une directamente a las caspasas, inhibiendo su actividad.
Sin embargo, también hay proteínas mitocondriales llamadas SMAC y DIABLO que inhiben los inhibidores. Al producirse una lesión mitocondrial se liberan y se unen a las IAP, liberando a las caspasas para que vayan a causar la apoptosis. Otro grupo de proteínas mitocondriales llamadas Htra2/Omi, el factor inductor de la apoptosis (AIF) y la endonucleasa G también pueden liberarse y escindir las IAP. El AIF también provoca la condensación de los cromosomas y la fragmentación del ADN independientemente de las caspasas.
De hecho, las mitocondrias son reguladores centrales de la apoptosis. Están implicadas las proteínas de la membrana mitocondrial externa Bcl-2, las proteínas de tipo BH3 y Bax: Bax puede formar un poro en la membrana para permitir que el citocromo c, normalmente situado en el espacio intermembrana, salga al citosol. Los monómeros de Bax se desplazan desde el citoplasma hasta la membrana mitocondrial externa, donde se oligomerizan y permiten la entrada de iones a través de la membrana. Esto también se ha demostrado en experimentos in vitro en los que se puede comprobar que las vesículas formadas por memrbanas mitocondriales externas se permeabilizan en presencia de Bax. Actualmente se desconoce por qué esta afluencia de iones conduce a la liberación del citocromo c.
Bcl-2 impide la liberación del citocromo c, bloqueando así la apoptosis. Bcl-2 fue el primer gen de apoptosis de mamíferos que se clonó. En algunos linfomas, se transloca a una posición bajo un promotor más fuerte, causando una sobreexpresión que impide que la célula cancerosa se apopte. Ver también mala & oferta.
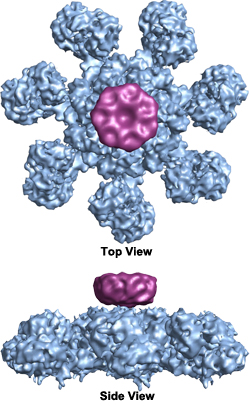
Una vez liberado el citocromo c, se une a Apaf-1 (factor activador de la proteasa apoptótica), haciendo que este último hidrolice el ATP al que suele estar unido, provocando así un cambio conformacional que activa a Apaf-1 y desencadena la cascada de caspasas. Apaf-1 forma un heptámero en forma de disco llamado «rueda de la muerte» o apoptosoma que activa las caspasas (Imagen de Wikimedia Commons por Org1012):
Cuando un factor trófico está presente, el receptor activa PI3K, que activa PKB/Akt, que fosforila Bad. El p-Bad es entonces retenido en el citosol por 14-3-3, impidiendo que el p-Bad inhiba a Bcl-2. Así se evita la apoptosis.
Los factores tróficos son un ejemplo de señal extrínseca celular que promueve la supervivencia. También hay señales extrínsecas que promueven la muerte (esto es el asesinato celular). El factor de necrosis tumoral (TNF-alfa) es liberado por los macrófagos para desencadenar la muerte celular al unirse a los «receptores de muerte». Los receptores de muerte tienen un único dominio transmembrana. Deben trimerizarse para activar el FADD (dominio de muerte asociado a Fas). Estos sirven como adaptadores para la caspasa-8 y -10 y forman un complejo de señalización inductor de la muerte (DISC) que puede iniciar la cascada de caspasas. Aunque todo este proceso se origina de forma independiente de las mitocondrias, también puede activar (…) t-Bid, dando lugar también a una señal de apoptosis mitocondrial.
Las células pueden volverse resistentes a la muerte expresando receptores señuelo que sólo tienen el dominio de unión del «ligando de la muerte» y ningún dominio citosólico activo. Esto ocurre a veces con normalidad en las células animales, pero también es un truco que utilizan algunos virus: codifican proteínas receptoras señuelo para mantener a sus células huésped a salvo del ataque inmunitario.
El TNF-alfa suele promover la muerte, pero también puede promover la supervivencia en ciertos tipos de células activando el NF-κB. A veces las células utilizan receptores señuelo para promover una respuesta inflamatoria en lugar de la muerte.
El p53 es un regulador clave de la respuesta al daño del ADN y puede promover la reparación del ADN, la apoptosis o la detención del ciclo celular. Lo hace uniéndose a los promotores de los genes diana. Todavía no está claro qué determina cuándo p53 inducirá la detención del ciclo celular frente a la apoptosis.
Métodos experimentales
Las células apoptóticas presentan una firma química particular. Uno de ellos es que una endonucleasa escinde el ADN en fragmentos en las regiones de enlace entre los nucleosomas y los fragmentos resultantes forman una escalera cuando se corren en un gel. Otra es la tinción TUNEL (Terminal deoxynucleotide transferase dUTP Nick End Labeling). Se trata de añadir una enzima Tdt y un BrdU que la Tdt añadirá a los extremos del ADN escindido. Después de darle la oportunidad de hacerlo, se lava el exceso de BrdU y luego se utiliza un anticuerpo contra el BrdU. Otro método es que la fosfatidilserina (PS) se encuentra normalmente en la hoja citosólica de la membrana plasmática; durante la apoptosis, se desplaza a la hoja exoplásmica, donde sirve como señal para solicitar a otras células que fagociten la célula moribunda. Una proteína anexina V marcada con fluorescencia puede etiquetar la PS en el exterior de las células apoptóticas.
El ADN de doble cadena no puede atravesar la membrana plasmática de las células intactas, y eso significa células sanas y células apoptóticas. Si sale, es un signo de necrosis. Así que se puede teñir con anexina V para el PS exoplásmico y con 7-AAD para el dsDNA; las células apoptóticas son las que son positivas para la anexina V pero negativas para el 7-AAD.
Vídeo final
En resumen, este es un vídeo inquietante sobre la apoptosis:
Relevancia para la PrP
En general, en la naturaleza, las células mueren por apoptosis, necrosis o por autofagia (es decir, en este caso, siendo engullidas enteras por otras células). En realidad, no hay otras formas de morir. Una de las muchas cosas misteriosas de las enfermedades priónicas es cómo mueren las neuronas en el cerebro infectado por priones: no parece que sigan ninguna de estas vías. Aquí hay una cita de un excelente artículo reciente sobre los mecanismos tóxicos de la enfermedad priónica :
La escisión de la caspasa 12 se produjo a las 10wpi, tras el aumento de la expresión de CHOP… coincidiendo con el inicio de la pérdida neuronal… sin embargo, el mecanismo efector exacto de la muerte neuronal no está claro: no encontramos ni apoptosis, ni autofagia, ni necrosis en el examen de los cortes del hipocampo… y ni la deleción de Bax, ni la sobreexpresión de Bcl-2, ni la deficiencia de caspasa 12 son neuroprotectores en la enfermedad priónica.
Además de sus propias pruebas, Moreno cita el estudio de la infección por priones en modelos de ratón con Bax (una proteína proapoptótica) eliminada o Bcl-2 (una proteína antiapoptótica) sobreexpresada, dos formas diferentes de bloquear la apoptosis. Ninguno de estos modelos de ratón presentó ningún retraso o mejora de la enfermedad priónica. Otra proteína apoptótica, la Caspasa-12, sufre un procesamiento proteolítico durante la infección por priones, pero la supresión de la Caspasa-12 tampoco cambió el curso de la enfermedad priónica.